Инновационные лекарства от рассеянного склероза могут стать доступнее
Инновационные лекарства для пациентов с рассеянным склерозом необходимо включить в перечень жизненно необходимых (ЖНВЛП) и клинические рекомендации не позже одного года с момента регистрации препарата.
Таким предложением Минздраву дополнен окончательный вариант решения, подготовленного по итогам Экспертного совета по здравоохранению при Комитете Совета Федерации по социальной политике. Документ есть в распоряжении «Парламентской газеты».
Чем опасен рассеянный склерозРассеянный склероз — одно из самых распространённых неврологических расстройств во всём мире, отметила в разговоре с «Парламентской газетой» заместитель председателя Комитета Совета Федерации по социальной политике, заслуженный врач России Татьяна Кусайко. По данным Общероссийской общественной организации инвалидов-больных рассеянным склерозом, в России до 150 тысяч пациентов с таким недугом. При этом выявить его непросто. Примерно у трети больных до постановки диагноза проходит пять лет.
При этом выявить его непросто. Примерно у трети больных до постановки диагноза проходит пять лет.
Сенатор напомнила, что власти предпринимают меры, призванные поддержать пациентов с этим заболеванием. С 2007 года оно вошло в перечень высокозатратных нозологий, и государство взяло на себя обязательство обеспечивать больных препаратами, изменяющими течение болезни. В регионах создают центры специализированной медицинской помощи больным рассеянным склерозом.
«Однако болезнь трансформируется, выявляются новые агрессивные формы заболевания, требующие внедрения новых подходов к лечению», — отметила Татьяна Кусайко. В ближайшие годы эксперты ожидают появления большого количества инновационных препаратов, в связи с этим врачебное сообщество считает необходимым обновить клинические рекомендации и изменить стандарты оказания медицинской помощи, рассказала парламентарий.
Также, по мнению сенатора, нужно подумать об увеличении финансирования на данную нозологию. «Внезапность возникновения рассеянного склероза, а также — зачастую — появление его в молодом возрасте, когда человек только получил образование, начал работать, завёл семью, — важные факторы, демонстрирующии необходимость создания программ адаптации в обществе пациентов с рассеянным склерозом», — добавила законодатель.
«Внезапность возникновения рассеянного склероза, а также — зачастую — появление его в молодом возрасте, когда человек только получил образование, начал работать, завёл семью, — важные факторы, демонстрирующии необходимость создания программ адаптации в обществе пациентов с рассеянным склерозом», — добавила законодатель.
Читайте также:
• В Совфеде предложили способы медподдержки пациентов с рассеянным склерозом • В России фиксируют дефицит жизненно важного препарата • В России с 2021 года увеличат производство онкопрепаратов
Также законодатели предлагают подумать о создании центров рассеянного склероза при ведущих НМИЦ и разработать стандарт специализированной помощи при обострении болезни.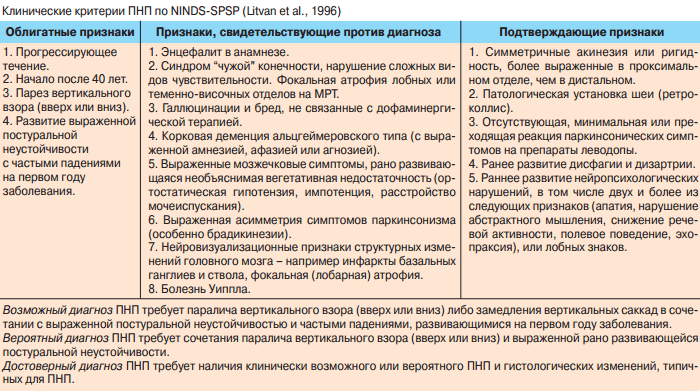 В программы дополнительного профессионального образования и ординатуры парламентарии считают необходимым включить вопросы профилактики, диагностики, лечения и реабилитации пациентов с этой болезнью. Кроме того, по их мнению, нужно разработать программу обучения неврологов амбулаторного звена по профилю рассеянный склероз.
В программы дополнительного профессионального образования и ординатуры парламентарии считают необходимым включить вопросы профилактики, диагностики, лечения и реабилитации пациентов с этой болезнью. Кроме того, по их мнению, нужно разработать программу обучения неврологов амбулаторного звена по профилю рассеянный склероз.
Минздраву, Минтруду и Минфину сенаторы предложили при проработке нового порядка по реабилитации учесть этапы лечения пациентов с рассеянным склерозом, а также воплощения в жизнь программ адаптации в обществе больных с таким диагнозом.
Дифференциальная диагностика атипичного паркинсонизма* — Центр Экстрапирамидных Заболеваний
Дифференциальная диагностика атипичного паркинсонизма*
О.С.Левин, Н.А.Амосова
Что такое атипичный паркинсонизм?
Атипичный паркинсонизм (АП) – условный термин, применяющийся для обозначения паркинсонического синдрома, отличающегося по своим характеристикам от синдрома, наблюдающегося при болезни Паркинсона (БП). Диагностика БП, являющейся причиной более чем 80% случаев паркинсонизма, в большинстве случаев не вызывает затруднений и может быть основана на клинических данных. Основными позитивными критериями диагноза БП могут служить: односторонние или асимметричные симптомы в дебюте заболевания, наличие типичного тремора покоя, хорошая реакция на препараты леводопы, относительно медленно прогрессирующее течение. К негативным критериям, подтверждающим диагноз БП, относится отсутствие пирамидных, мозжечковых знаков, грубого псевдобульбарного синдрома, раннего развития постуральной неустойчивости, тяжелой вегетативной недостаточности, надъядерной офтальмоплегии и некоторых других симптомов. Соответственно, атипичным паркинсонизмом мы можем назвать акинетико-ригидный синдром, при котором препараты леводопы не оказывают эффекта или оказывают минимальный или краткосрочный эффект, отсутствует тремор покоя, отмечается двустороннее относительно симметричное начало, имеются сопутствующие симптомы, не свойственные БП (см.
Диагностика БП, являющейся причиной более чем 80% случаев паркинсонизма, в большинстве случаев не вызывает затруднений и может быть основана на клинических данных. Основными позитивными критериями диагноза БП могут служить: односторонние или асимметричные симптомы в дебюте заболевания, наличие типичного тремора покоя, хорошая реакция на препараты леводопы, относительно медленно прогрессирующее течение. К негативным критериям, подтверждающим диагноз БП, относится отсутствие пирамидных, мозжечковых знаков, грубого псевдобульбарного синдрома, раннего развития постуральной неустойчивости, тяжелой вегетативной недостаточности, надъядерной офтальмоплегии и некоторых других симптомов. Соответственно, атипичным паркинсонизмом мы можем назвать акинетико-ригидный синдром, при котором препараты леводопы не оказывают эффекта или оказывают минимальный или краткосрочный эффект, отсутствует тремор покоя, отмечается двустороннее относительно симметричное начало, имеются сопутствующие симптомы, не свойственные БП (см.
Причиной АП может быть целый ряд заболеваний, при которых перечисленные симптомы встречаются в той или иной комбинации. Поэтому АП – условный термин, который обычно используют до того момента, когда с помощью клинического и параклинического обследования, а иногда и длительного наблюдения удастся поставить нозологический диагноз.
В Центре экстрапирамидных заболеваний за последние 2 года наблюдалось 98 больных с АП. В результате обследования клинический диагноз удалось установить более чем у 3/4 из них, у 22 больных он остается неясным (табл. 2).
Таблица 1. Основные клинические черты АП
| Основные группы признаков | Признаки |
| Особенности паркинсонического синдрома | Отсутствие значительного и стойкого эффекта препаратов леводопы Отсутствие тремора покоя Симметричные проявления Быстрое прогрессирование Раннее развитие постуральной неустойчивости и падений |
| Сопутствующие неврологические синдромы | Выраженный постурально-кинетический тремор и миоклония Раннее развитие выраженной вегетативной недостаточности Ограничение подвижности глазных яблок Раннее развитие тяжелых псевдобульбарных симптомов (дизартрия, дисфагия) Пирамидные знаки Аксиальная дистония Раннее развитие деменции; наличие очаговых нарушений корковых функций (апраксия, афазия и др. |
 )
)Таблица 2. Заболевания, проявляющиеся синдромом АП
| Заболевания | Процент от общего числа больных |
| Деменция с тельцами Леви | 20 |
| Мультисистемная атрофия | 15 |
| Прогрессирующий надъядерный паралич | 12 |
| Сосудистый паркинсонизм | 11 |
| Лекарственный паркинсонизм | 5 |
| Кортикобазальная дегенерация | 3 |
| Токсический паркинсонизм | 3 |
| Гепатолентикулярная дегенерация | 3 |
| Нормотензивная гидроцефалия | 2 |
| Болезнь Фара | 2 |
| Опухоль мозга | 1 |
| Болезнь Крейтцфельдта–Якоба | 1 |
| Нозологический диагноз неясен | 22 |
| Всего | 100 |
Основной причиной АП являются нейродегенеративные заболевания, которые обычно относят к «паркинсонизму-плюс»: мультисистемная атрофия (МСА), прогрессирующий надъядерный паралич (ПНП), кортикобазальная дегенерация (КБД), болезнь диффузных телец Леви (БДТЛ). Реже причиной АП бывают болезни, вызывающие вторичный паркинсонизм (сосудистый, лекарственный, токсический паркинсонизм, нормотензивная гидроцефалия, опухоли, глиоматоз мозга). Нельзя исключить, что отдельные случаи АП представляют собой вариант БП с нетипичными проявлениями или необычным течением.
Реже причиной АП бывают болезни, вызывающие вторичный паркинсонизм (сосудистый, лекарственный, токсический паркинсонизм, нормотензивная гидроцефалия, опухоли, глиоматоз мозга). Нельзя исключить, что отдельные случаи АП представляют собой вариант БП с нетипичными проявлениями или необычным течением.
Ограниченность объема статьи заставляет нас остановиться лишь на некоторых неврологических, нейропсихологических и нейровизуализационных признаках, имеющих, по нашему опыту, важное значение в дифференциальной диагностике наиболее частых заболеваний, вызывающих АП.
Стойкая высокая эффективность препаратов леводопы – наиболее надежный признак БП, отмечающийся в 95% случаев. Таким образом, назначение леводопы можно рассматривать как своеобразный фармакологический тест, позволяющий оценить состояние как нигростриарных нейронов, так и, главным образом, стриарных нейронов, несущих дофаминовые рецепторы. Практически всем заболеваниям, бывающим причиной АП, свойственна низкая эффективность препаратов леводопы, однако из этого правила есть исключения.
 Таким образом, низкая эффективность леводопы – важный признак АП, но и хорошая реакция на леводопу не исключает его. Развитие психотических нарушений при приеме препаратов леводопы наиболее характерно для БДТЛ, а на более позднем этапе возможно и при БП, в то же время они крайне редко наблюдаются при ПНП, МСА и КБД.
Таким образом, низкая эффективность леводопы – важный признак АП, но и хорошая реакция на леводопу не исключает его. Развитие психотических нарушений при приеме препаратов леводопы наиболее характерно для БДТЛ, а на более позднем этапе возможно и при БП, в то же время они крайне редко наблюдаются при ПНП, МСА и КБД.Важное диагностическое значение имеет и другой фактор. При длительном лечении препаратами леводопы, обычно через 3–5 лет, у пациентов с БП закономерно возникают моторные флуктуации и дискинезии. При заболеваниях, проявляющихся АП, эти феномены отсутствуют или имеют своеобразный характер. Например, при МСА крайне редко развиваются хореодистонические дискинезии в конечностях, возникающие при БП в связи с приемом леводопы, но нередко на фоне приема препаратов леводопы возникают дискинезии краниоцервикальной локализации. Мы неоднократно наблюдали у больных МСА дистоническую камптокормию (выраженное сгибание туловища кпереди) в связи с применением дофаминергических средств. Следует подчеркнуть, что вывод о неэффективности препарата леводопы может быть сделан лишь после того, как доза леводопы (в составе комбинированного препарата, включающего ингибитор ДОФА-декарбоксилазы) будет доведена до 1500 мг/сут. Если в течение 2 нед. и эта доза окажется неэффективной, то ее следует постепенно снизить до средней (500–750 мг/сут.). Полная отмена препаратов леводопы, даже если они казались неэффективными, нежелательна, так как может привести к ухудшению состояния больного. В первую очередь это относится к больным с МСА. У части больных МСА наблюдается раннее (на 1–2-м году заболевания) развитие моторных флуктуаций.
Симметричность паркинсонических симптомов. У большинства пациентов с БП тремор, акинезия или ригидность первоначально вовлекают только одну конечность или сторону тела (гемипаркинсонизм). При последующем прогрессировании симптомы появляются и с другой стороны, но асимметрия часто сохраняется, и конечности, вовлеченные первыми, оказываются более пораженными. Одностороннее начало и выраженная асимметрия акинезии, ригидность, постурально-кинетический тремор характерны и для КБД. Но для большинства заболеваний, проявляющихся синдромом АП, свойственно двустороннее начало и более выраженная симметричность симптомов. Относительная симметричность паркинсонических симптомов особенно характерна для ПНП, в несколько меньшей степени – для СП. Хотя при МСА и БДТЛ симптомы нередко бывают симметричными, у значительной части больных возможно асимметричное вовлечение конечностей.
Распределение паркинсонических симптомов. При БП в первую очередь чаще всего вовлекаются дистальные отделы конечностей. При ПНП и СП акинезия и ригидность более выражены в аксиальных отделах (голова, шея, туловище) и проксимальных отделах конечностей, тогда как в дистальных отделах признаки гипокинезии могут отсутствовать, а мышечный тонус бывает нормальным, а иногда и низким. По отношению к этому признаку МСА и ДТЛ занимают промежуточное положение: у некоторых больных доминируют аксиальные симптомы, у других аксиальные и дистальные симптомы бывают выражены в одинаковой степени.
Постуральные нарушения и изменения ходьбы. Для большинства заболеваний, проявляющихся синдромом АП, характерно гораздо более быстрое, чем при БП, развитие постуральной неустойчивости, которая нередко превращается в основной инвалидизирующий фактор, резко затрудняющий ходьбу и часто приводящий к падениям и травмам. Особенно быстро нарастает постуральная неустойчивость при ПНП, СП и нормотензивной гидроцефалии. Для проверки постуральной устойчивости врач становится за спиной больного и быстрым толчком кзади выводит его из равновесия. При неэффективности постуральных рефлексов у больного возникает ретропульсия либо он может упасть как «подпиленное дерево», даже не предпринимая попыток удержаться на ногах.
Важное дифференциально-диагностическое значение имеет и оценка ходьбы. У больных с СП, МСА, БДТЛ, КБД чаще развиваются нарушения ходьбы по типу лобной дисбазии (апраксии ходьбы). Для лобной дисбазии характерны замедленное начало ходьбы, походка мелкими шаркающими шажками, затруднения при поворотах и преодолении даже небольших препятствий, часто с застыванием и топтанием. Но в отличие от БП площадь опоры при ходьбе не уменьшается, менее выражены сгибательная поза и ахейрокинез (следует оговорить, что у некоторых больных с МСА и БДТЛ походка такая же, как и при БП, а у больных с поздней стадией БП нарушение ходьбы может приобретать черты лобной дисбазии).
У части больных с ПНП походка также меняется по типу лобной дисбазии, но в целом для этого заболевания более характерны изменения ходьбы по типу подкорковой астазии. На ранней стадии у таких больных не отмечается затруднений инициации ходьбы, уменьшения длины шага и площади опоры, снижения скорости ходьбы, как при БП. Затруднения больного скорее связаны с тем, что он не в состоянии правильно скоординировать движения туловища и нижних конечностей таким образом, чтобы не происходило резких смещений центра тяжести тела относительно площади его опоры. Голова и туловище больных нередко бывают отклонены кзади. Из-за размашистых шагов ноги время от времени опережают «задержавшееся» туловище, и больной может неожиданно упасть на спину, даже не предпринимая попыток удержать равновесие. Падения при ПНП могут происходить в любом направлении, но чаще больной валится на спину (при БП и МСА больные чаще падают вперед). При МСА ноги у некоторых больных бывают широко расставленными, а ходьба имеет более отчетливый атактический характер.
Пирамидные знаки исключают БП, но нередко отмечаются при различных заболеваниях, проявляющихся АП, чаще всего при СП и МСА. Следует, однако, отметить, что интерпретировать оживление сухожильных рефлексов в отсутствие патологических стопных знаков, расширения рефлексогенных зон или клонуса стоп как проявление пирамидной недостаточности следует с осторожностью. Поводом для ошибочной диагностики пирамидной недостаточности у больных с экстрапирамидной патологией нередко является особый вариант дистонии стопы («стриарная стопа»), для которой характерна разгибательная установка большого пальца. В отличие от рефлекса Бабинского, разгибание большого пальца в этом случае представляет собой не фазический, а тонический феномен, часто возникающий спонтанно или провоцирующийся ходьбой и другими движениями. Штриховое раздражение подошвы при «стриарной стопе» вызывает нормальную сгибательную реакцию стопы (обычно ее легче вызвать, если придать ноге положение легкой флексии).
Глазодвигательные нарушения. Подробное исследование глазодвигательных функций – обязательная часть осмотра у больных с АП, особенно важная для диагностики ПНП. Для этого заболевания характерно ограничение амплитуды содружественных движений глаз (паралич взора) вниз, но в значительной части случаев оно развивается лишь спустя 2–3 года от начала болезни. Но еще до развития паралича взора у большинства больных с ПНП выявляются более легкие глазодвигательные нарушения: замедление и гипометрия вертикальных саккадических движений и нарушение плавности вертикальных следящих движений (особенно направленных вниз), а также нарушение подавления вестибулоокулярного рефлекса и отсутствие или замедление быстрой фазы оптико-кинетического нистагма в вертикальной плоскости.
Для проверки произвольных саккадических движений врач просит больного быстро переводить взгляд с одной точки на другую в пределах поля зрения (например, с одного пальца врача на другой, при этом пальцы обычно разводят на 1 м и помещают на расстоянии 60 см от уровня глаз больного). Уже на ранней стадии ПНП нередко можно отметить, что вертикальные саккады оказываются диспропорционально более медленными, чем горизонтальные, а из-за гипометрии вертикальных саккад для того, чтобы перевести взор с одной точки на другую, глаза совершают одно или несколько дополнительных движений. Неспособность подавить вестибулоокулярный рефлекс можно проверить, приставив ко лбу больного молоток и попросив его фиксировать взгляд на рукоятке молотка: при повороте головы взгляд больного будет время от времени отставать от мишени, а затем догонять ее быстрым скачком. Оптико-кинетический нистагм проверяют, вращая перед глазами больного барабан с чередующимися черными и белыми полосами. У некоторых больных с ПНП, часто уже на ранней стадии, нарушается фиксация взора: если попросить больного остановить взгляд на удаленной точке, то можно заметить внезапные очень быстрые скачкообразные подергивания глазных яблок от точки фиксации и обратно, обычно в пределах 1–5° (дисфиксационные саккады). Для КБД характерен длительный латентный период саккадических движений в горизонтальной плоскости.
Умеренные глазодвигательные нарушения, в том числе дисфиксационные саккады, возможны и при других заболеваниях, проявляющихся синдромом паркинсонизма (табл. 3). Важно предостеречь от диагностики ПНП только на основании выявления у больного с паркинсонизмом пареза взора вверх: это неспецифический признак, который может наблюдаться практически при любом варианте АП.
Кроме того, следует отметить, что и парез взора вниз может наблюдаться не только при ПНП, но также при КБД и БДТЛ. Характерной чертой ПНП являются исключительная редкость мигания, нередко парадоксально сочетающаяся с резким оживлением надбровного рефлекса. Иногда больные с ПНП испытывают затруднения при произвольном открывании, реже закрывании глаз (апраксия открывания или закрывания глаз). Изредка аналогичные расстройства наблюдаются при КБД. Хотя блефароспазм чаще встречается при ПНП, его дифференциально-диагностическое значение невелико, так как он возможен при БП и других формах АП.
Псевдобульбарный синдром. Раннее развитие псевдобульбарных проявлений, особенно дизартрии и дисфагии, в большей степени характерно
Таблица 3. Сравнительная частота различных форм глазодвигательных нарушений при заболеваниях, проявляющихся синдромом паркинсонизма
| Тип нарушения | БП | МСА | ПНП | КБД | БДТЛ |
| Парез взора | |||||
| вертикального | |||||
| – вверх | + | + | +++ | ++ | ++ |
| – вниз | – | – | +++ | + | + |
| горизонтального | – | ++ | ++ | ++ | – |
| Нарушение плавности следящих движений | + | ++ | +++ | ++ | + |
| Гипометрия саккад | + | ++ | ++ | ++ | + |
| Замедление саккад | + | ++ | +++ | ++ | + |
| Гиперметрия глазных яблок | – | + | – | – | – |
| Дисфиксационные саккады | – | +++ | +++ | ++ | + |
| Отсутствие феномена Белла | – | – | +++ | ++ | – |
| Нистагм | – | ++ | + | – | – |
| Апраксия открывания/закрывания глаз | – | – | +++ | ++ | – |
для ПНП, СП, МСА. В отличие от БП, при которой дизартрия в основном вызвана гипокинезией и характеризуется монотонной приглушенной речью, при ПНП и СП доминирует спастический компонент дизартрии, проявляющийся охриплостью и огрубением голоса, замедленной, растянутой, смазанной речью, прерываемой частыми паузами. При МСА спастический компонент выражен меньше и в части случаев преобладает атактическая скандированная речь. У части больных с МСА развивается паралич мышц гортани, вызывающий инспираторный стридор и своеобразную дисфонию. Особенности дрожательного гиперкинеза. У 85% пациентов с БП выявляется характерный тремор покоя по типу «скатывания пилюль», который обычно бывает асимметричным и иногда сопровождается умеренным постурально-кинетическим тремором. При СП тремор покоя выявляется только в единичных случаях, связанных с поражением среднего мозга. Тремор покоя не характерен и для ПНП (только у 5% пациентов с этим заболеванием наблюдается минимальный тремор покоя). Чаще тремор покоя встречается при МСА и БДТЛ, но при этом у большинства больных с МСА и половины больных с БДТЛ он отсутствует. При СП, ПНП, БДТЛ нередко встречается умеренный постурально-кинетический тремор. Грубый постурально-кинетический тремор с интенционным компонентом, на который иногда наслаиваются акционные миоклонические подергивания, наблюдается при КБД и иногда при МСА. При МСА этот тремор обычно бывает двусторонним, при КБД – односторонним или резко асимметричным.
Другие экстрапирамидные синдромы. При БП возможно развитие дистонии (чаще всего дистонии стопы, усиливающейся при ходьбе), но в большинстве случаев дискинезии бывают связаны с применением леводопы. При АП наблюдается более широкий спектр других экстрапирамидных синдромов. Особенно часто встречается аксиальная дистония: при ПНП – ретроколлис и блефароспазм, при МСА – антероколлис. Дистония конечностей часто наблюдается при КБД, а при ПНП она бывает практически единственным асимметричным проявлением заболевания. У части больных с МСА, КБД, БДТЛ наблюдается акционная миоклония.
Вегетативная дисфункция. Выраженные признаки вегетативной недостаточности: ортостатическая гипотензия с частыми предобморочными состояниями или обмороками, гипертензия в положении лежа, гипотензия после еды, фиксированный пульс, импотенция, учащенное или затрудненное мочеиспускание, нарушение моторики желудочно-кишечного тракта или расстройства потоотделения и т.д. – особенно характерны для МСА, но могут также наблюдаться при БП, БДТЛ, ПНП. Важное диагностическое значение имеет время появления симптомов; например, при МСА клинически явная вегетативная недостаточность часто выявляется в первые 1–3 года болезни, тогда как при БП – спустя 5 лет от момента появления первых симптомов. При КБД признаки вегетативной недостаточности обычно отсутствуют.
Особенности течения заболевания. Для большинства заболеваний, вызывающих АП, характерно более быстрое прогрессирование, чем при БП. Более того, если через 6–8 лет после начала заболевания больной сохраняет способность к самостоятельному передвижению, то это свидетельствует против диагноза ПНП или МСА. Для нейродегенеративных заболеваний (ПНП, МСА, КБД) характерны постепенное начало и неуклонно прогрессирующее течение. Отличительными особенностями СП являются острое или подострое начало, ступенеобразное прогрессирование с периодами частичного регресса и длительной стабилизации.
Особенности нейропсихологических нарушений. Нейропсихологическое обследование является весьма информативным дополнением неврологического осмотра, так как позволяет лучше оценить топику и распространенность патологического процесса. При МСА, как и в большинстве случаев БП, когнитивные нарушения не достигают степени деменции и связаны с дисфункцией подкорково-лобных кругов. Они проявляются замедленностью мышления (брадифренией), нарушением внимания, снижением мотивации, трудностями в инициации, планировании и организации интеллектуальной деятельности с вторичной дефектностью мнестических процессов. При ПНП подкорково-лобный когнитивный дефект гораздо более выражен и достигает степени деменции, сопровождаясь выраженным изменением поведения по лобному типу (апатико-абулическим синдромом, расторможенностью, импульсивностью). Развитие деменции при ПНП может быть связано с массивной деафферентацией лобной коры, а на поздних этапах, возможно, с непосредственным поражением коры. При БДТЛ, как и у части пациентов с БП, развивается деменция смешанного подкорковокоркового типа, для которой характерна комбинация подкорково-лобного нейропсихологического дефекта с признаками вовлечения височно-теменной коры (грубым первичным нарушением памяти, афазией, апраксией, пространственной дезориентацией и т.д.). В отличие от БП с деменцией, при БДТЛ деменция развивается более быстро (в первые 1–2 года заболевания), сопровождается выраженными флуктуациями с развитием эпизодов инактивности или спутанности, продолжающихся от нескольких часов до нескольких суток, и ранним развитием психотических нарушений (зрительных галлюцинаций, параноидного синдрома). Ключевую роль играет нейропсихологическое исследование и в диагностике КБД, при которой оно выявляет, наряду с подкорково-лобным дефектом, признаки поражения лобной и теменной коры (чаще всего апраксию, афазию и игнорирование половины пространства).
Компьютерная томография (КТ) и особенно магнитно-резонансная томография (МРТ) имеют особенно важное значение в диагностике сосудистого и других форм вторичного паркинсонизма. В диагностике нейродегенеративных заболеваний роль нейровизуализации гораздо более ограниченна и сводится прежде всего к исключению структурной патологии мозга (опухоли, гидроцефалии, сосудистых поражений). В то же время МРТ может выявлять и некоторые позитивные признаки нейродегенеративных заболеваний, диагностический вес которых, однако, до настоящего времени мало изучен. Например, у части больных с МСА МРТ выявляет снижение интенсивности сигнала от скорлупы, связанное с накоплением железа (на Т2-взвешенных изображениях). Однако этот же признак может выявляться и у больных с ПНП. Более специфичны для МСА щелевидная гиперинтенсивная (в Т2-режиме) зона по наружному краю скорлупы и атрофия скорлупы. Кроме того, при этом заболевании нередко встречаются также атрофия моста и мозжечка, но этот признак иногда выявляется и при ПНП и КБД. Атрофия среднего мозга особенно характерна для ПНП, но возможна также при МСА и КБД. Важно учитывать, что при ПНП МРТ или КТ нередко выявляют лакунарные очаги в веществе мозга, в том числе и в базальных ганглиях, связанные с артериальной гипертензией, которая может развиваться в результате дегенерации стволовых вегетативных структур. Выявление таких очагов может быть причиной ошибочной диагностики СП у больных с ПНП. Чтобы избежать этой ошибки, нужно оценивать весь комплекс нейровизуализационных изменений и всегда соотносить их с клиническими проявлениями.
Некоторое диагностическое значение имеет и региональное распределение наружной атрофии, выражающейся в расширении корковых борозд. Выраженная наружная атрофия мозга, преимущественно в лобных и височных отделах, чаще всего встречается при БДТЛ, но возможна при ПНП и БП. При КБД МРТ может выявлять асимметричную атрофию, преимущественно вовлекающую лобную и теменную кору полушария, контралатерального наиболее пораженной руке. У большинства больных с МСА величина корковых борозд и желудочковой системы соответствует возрастной норме.
Таблица 4. Основные диагностические признаки заболеваний, являющихся причиной АП
| Болезни | Обязательные признаки | Признаки, свидетельствующие в пользу диагноза | Признаки, свидетельствующие против диагноза |
| ПНП | Акинезия и ригидность в аксиальных отделах, выраженная постуральная неустойчивость с частыми падениями, парез вертикального взора | Выраженный псевдобульбарный синдром, ретроколлис, подкорковая астазия, деменция подкорково-лобного типа. Атрофия среднего мозга при МРТ | Высокая эффективность препаратов леводопы, длительное доброкачественное течение (возможность самостоятельного передвижения через 5 лет), асимметрия клинических проявлений, корковые черты деменции, корковые нарушения чувствительности, галлюцинации |
| МСА | Акинезия, ригидность (паркинсоническая) или мозжечковая атаксия с рано развивающейся вегетативной недостаточностью | Резистентность к препаратам леводопы, пирамидные знаки, антероколлис, псевдобульбарные нарушения, парез мышц гортани, лицевая дискинезия при применении леводопы, ранние моторные флуктуации, миоклония. Изменения интенсивности сигнала от скорлупы, атрофия моста и мозжечка при МРТ | Стойкая высокая эффективность препаратов леводопы; длительное доброкачественное течение; деменция, спутанность сознания и галлюцинации, связанные с приемом противопаркинсонических средств |
| БДТЛ | Акинезия, ригидность, деменция | Флуктуации психического статуса, психотические нарушения, вегетативная недостаточность. Атрофия лобных и височных долей при МРТ. Вспышки медленных волн в лобной области при ЭЭГ | Отсутствие деменции в первые 2 года заболевания, отсутствие признаков корковой деменции |
| КБД | Асимметричные акинезия, ригидность, дистония; очаговые нарушения высших корковых функций (апраксия, афазия, игнорирование половины пространства) | Феномен «чужой» руки, астерео гноз, грубые асимметричные постурально-кинетический тремор и миоклония. Асимметричная атрофия лобной и теменной коры при МРТ | Раннее развитие деменции, высокая эффективность леводопы |
| Болезни | Обязательные признаки | Признаки, свидетельствующие в пользу диагноза | Признаки, свидетельствующие против диагноза |
| СП | Акинезия, ригидность, признаки цереброваскулярного заболевания | Подострое начало, ступенеобразное прогрессирование с периодами регресса и длительной стабилизации, низкая эффективность леводопы, паркинсонические признаки преимущественно в аксиальных отделах и нижних конечностях, раннее развитие постуральной неустойчивости и тазовых нарушений, лобная дисбазия, выраженный псевдобульбарный синдром. Обширный лейкоареоз или двусторонние подкорковые инфаркты в скорлупе и бледном шаре при МРТ | Высокая стойкая эффективность препаратов леводопы, отсутствие очаговых или диффузных изменений при МРТ предположительно сосудистого характера |
ПНП – прогрессирующий надъядерный паралич, МСА – мультисистемная атрофия, БДТЛ – болезнь диффузных телец Леви, СП – сосудистый паркинсонизм, КБД – кортикобазальная дегенерация.
В заключение следует отметить, что клинические признаки заболеваний, вызывающих синдром АП, в значительной степени перекрываются в связи с тем, что в патологический процесс при различных процессах вовлекаются одни и те же структуры. В то же время особенности пространственно-временны́ х характеристик того или иного патологического процесса, выражающиеся в относительно специфической констелляции поражаемых структур (систем) или, что, быть может, еще более важно, в определенной последовательности их вовлечения, предопределяют клиническое своеобразие каждого из заболеваний и создают возможность установления у больных с АП нозологического диагноза. Наш опыт показывает, что в большинстве случаев АП диагноз может быть установлен при учете всего комплекса неврологических, нейропсихологических и нейровизуализационных признаков, в котором находит отражение патофизиологическая логика процесса, специфичная у каждого из рассмотренных заболеваний. Основные дифференциально-диагностические признаки заболеваний, вызывающих АП, суммированы в таблице 4. Важно подчеркнуть, что клинический диагноз заболевания, вызывающего АП, нередко носит вероятностный характер и может быть пересмотрен при последующем наблюдении в связи с появлением новых признаков.
* Диагностика и лечение экстрапирамидных расстройств / Под ред. В.Н.Штока. – М., 2000. – С. 71–83.
дисметрия — это… Что такое дисметрия?
ДИСМЕТРИЯ — ДИСМЕТРИЯ, отсутствие меры в движении как в пространственном, так и во времен нбм отношении: движение переходит поставленную цель, останавливается слишком поздно, выполняется порывисто, с излишней Чрезмерное открытие правой и левой руки при… … Большая медицинская энциклопедия
дисметрия — [дис… + гр. мера] – мед. отсутствие в движениях меры; симптом поражения мозжечка Большой словарь иностранных слов. Издательство «ИДДК», 2007 … Словарь иностранных слов русского языка
Дисметрия — (дис + греч. metron – мера, размер). Нарушение координации движений вследствие утраты чувства расстояния, соразмерности и точности двигательных актов. Движения больных становятся размашистыми, увеличиваются в объеме, недостаточно точны,… … Толковый словарь психиатрических терминов
дисметрия — (от. греч. dis приставка, означающая расстройство, и metron мера, размер) нарушение координации движений вследствие утраты чувства расстояния, соразмерности и точности двигательных актов. Движения становятся размашистыми, увеличиваются в… … Дефектология. Словарь-справочник
МОЗЖЕЧКОВО-МОСТОВОЙ УГОЛ — (Klein hirnbruckenwinkel, angle ponto cerebelleuse, по нек рым angle ponto bulbo cerebelleuse) занимает своеобразное место в невропатологии, неврогистопатологии и неврохирургии. Названием этим обозначается угол между мозжечком, продолговатым… … Большая медицинская энциклопедия
ДИССИНЕРГИЯ — ДИССИНЕРГИЯ, dyssynergia (от греч. dys приставка, означающая качественное расстройство, syn с, вместе и ergon работа), расстройство синергии движений в смысле нарушения правильной последовательности и сочетания действий; чаще всего мозжечкового… … Большая медицинская энциклопедия
Алкого́льное опьяне́ние — патологическое состояние, характеризующееся сочетанием психических и неврологических расстройств, обусловленных воздействием этилового спирта на центральную нервную систему. Алкоголь, легко растворяясь в липидах, оказывает мембранотоксическое… … Медицинская энциклопедия
Альтерни́рующие синдро́мы — симптомокомплексы, характеризующиеся сочетанием поражения черепных нервов на стороне очага с проводниковыми нарушениями движения и чувствительности на противоположной стороне. Возникают при поражении одной половины ствола головного мозга,… … Медицинская энциклопедия
Движе́ния — являются формой взаимодействия организма с окружающей средой, осуществляющейся благодаря регуляторным физиологическим механизмам, основные уровни которых находятся в ц.н.с. Движения происходят с помощью поперечнополосатых мышц, их… … Медицинская энциклопедия
Русси́ — Леви́ синдро́м — (G. Roussy, франц. патологоанатом, 1874 1948; G. Levy, франц. невролог, 1886 1935; синоним гередитарная арефлекторная дистазия) комплекс наследственных неврологических симптомов, включающий проявления полиневропатии, атаксию и характерную… … Медицинская энциклопедия
Мультисистемная атрофия или прогрессирующий надъядерный паралич?
Введение. Мультисистемная атрофия (МСА) и прогрессирующий надъядерный паралич (ПНП) – нейродегенеративные заболевания, которые раньше относились к группе болезней «паркинсонизм-плюс». В основе клинической картины – синдром Паркинсона с низкой эффективностью специфической терапии, когнитивные нарушения и дополнительные симптомы, на основании которых в дальнейшем и были определены отдельные нозологии [1]. И при МСА, и при ПНП поражаются подкорковые и стволовые структуры.
Оба заболевания, мультисистемная атрофия и прогрессирующий надъядерный паралич, встречаются в неврологической практике в основном как спорадические; мультисистемная атрофия имеет несколько типов развития и течения болезни, которые могут быть представлены как «чистыми», так и «смешанными» вариантами [2], для прогрессирующего надъядерного паралича также характерны различные клинические варианты дебюта, развития заболевания, описаны как традиционные, так и необычные и нетипичные сочетания симптомов [3,4], что приводит к существенным трудностям в диагностике каждого из этих заболеваний. На ранних стадиях, когда симптомы выражены еще неярко, а клинические синдромы находятся в процессе формирования, данные заболевания имеют значительное количество общих черт в виде синдрома паркинсонизма, вегетативной недостаточности, глазодвигательных нарушений, псевдобульбарного синдрома, когнитивных нарушений, эмоционально-личностных расстройств и нарушений сна, что вызывает существенные трудности в проведении дифференциальной диагностики.
В настоящее время проблема дифференциальной диагностики данных болезней представляет интерес как из-за различного прогноза в плане развития деменции и сохранения самостоятельности пациента [4], так и из-за выявления семейных случаев и при мультисистемной атрофии, и при прогрессирующем надъядерном параличе, в связи с чем предполагается, что генетические факторы могут влиять на патогенез и развитие этих заболеваний. По данным молекулярно-генетических исследований, один из вариантов мультисистемной атрофии связан с мутацией в гене COQ2, расположенном в области 4q21.22-q21.23 [5], семейный вариант прогрессирующего надъядерного паралича – с мутацией в гене МАРТ, расположенном в области 17q21.31 [6].
Описание клинического случая. В качестве иллюстрации сложности проведения дифференциального диагноза между мультисистемной атрофией и прогрессирующим надъядерным параличом приводим описание клинического случая динамического наблюдения за пациенткой М., 60 лет, несколько раз проходившей лечение в различных отделениях Воронежской городской клинической больницы скорой медицинской помощи № 1.
Впервые в поле зрения врача-невролога пациентка М. попала, когда находилась на лечении в гинекологическом отделении ВГКБМСП № 1 по поводу вульвовагинита и вульводинии. К неврологу была направлена на консультацию.
Жалобы и история развития заболевания.
Пациентку беспокоили периодические головные боли, эпизоды повышения АД до 150/90, ощущение перебоев в работе сердца, ухудшение зрения (с -5 диоптрий до -9 за последние 3 года). В качестве основной жалобы предъявляла неустойчивость при ходьбе, частые падения, особенно в сырую слякотную погоду. При падении ушибается, получает ссадины. Сознания не теряет. Дважды больная «предчувствовала» падения: шла по улице и поняла, что сейчас упадет. Попыталась этого не допустить, однако все равно упала. Случайный прохожий, помогавший подняться, заметил: «От чего же вы упали, ведь даже не споткнулись?!». Несколько лет назад был эпизод, когда больная шла по улице, почувствовала шаткость при ходьбе и резкое отклонение в сторону, «стало уводить». Пациентка попыталась «откорректировать курс», однако это не удалось сделать, поэтому, чтобы не выйти на проезжую часть, ей пришлось взяться за столб и некоторое время за него держаться. Затем состояние улучшилось, больная смогла продолжить путь. После установления в процессе беседы доверительных отношений с врачом, пациентка призналась, что помимо неустойчивости и падений ее беспокоит учащенное мочеиспускание, иногда доходящее до нескольких раз в час, с императивным характером позывов, нарушение в связи с частым мочеиспусканием ночного сна, трудности в общении, профессиональной деятельности. Беспокоит учащенная дефекация (часто вместе с мочеиспусканием), жидкий стул. В настоящее время помимо учащенного мочеиспускания жалуется на покалывание и жжение в области наружных половых органов при мочеиспускании, связывает это с обострением вульвовагинита.
Пациентка считает, что здоровье ее нарушилось около 3-х лет назад, когда больная самостоятельно выдавила и прижгла бриллиантовой зеленью фурункул во влагалище. Вскоре после этого появились дизурические жалобы: неприятные ощущения при мочеиспускании, ощущение, что в мочеиспускательном канале есть какое-то образование, инородное тело или воспалительный инфильтрат. Больная отметила также более частые позывы на мочеиспускание, акт мочеиспускания периодически бывал болезненным. Сначала лечилась самостоятельно «от цистита», принимала различные антибиотики, затем обращалась к урологам, был диагностирован острый, затем хронический цистит, который часто обострялся, что подтверждалось анализами мочи, ультразвуковым исследованием мочевого пузыря. Пациентка в течение последних двух лет по поводу гиперактивного мочевого пузыря по рекомендации урологов принимает препарат спазмекс. Начинала прием с минимальных доз, препарат давал положительный эффект, но периодически все равно приходилось увеличивать дозу лекарственного средства. В настоящее время пациентка отмечает существенное снижение эффективности препарата, особенно в течение последних нескольких месяцев, недель. Для поездок, работы пользуется прокладками, при прогулках по городу строит маршруты так, чтобы можно было зайти в какое-либо учреждение в туалет. Пациентка отмечает, что все позывы на мочеиспускание сильные, прохождение мочи по мочеиспускательному каналу чувствует хорошо. Количество принимаемой жидкости оценивает как небольшое или среднее, около 1,5-2 литров. Жажду отрицает.
Анамнез жизни.
Работает юристом, разведена, проживает с сыном, страдающим психическим заболеванием. У пациентки также есть дочь, живущая отдельно со своей семьей. Несколько лет назад был вывих ключицы в автоаварии, восстановилась быстро и хорошо, вернулась к работе. Онкологические, венерические заболевания, туберкулез, болезнь Боткина, отрицает. В течение ряда лет отмечались аллергические реакции на пыльцу растений. На продукты питания, медикаменты аллергию отрицает. Наблюдалась у ЛОР-врача с диагнозом аллергический ринит.
Неврологический статус. Больная в ясном сознании, контактна, адекватна, правильно ориентирована. Обращает на себя внимание телосложение и походка: пациентка астенического телосложения, высокая, плечи развернуты назад, голова также несколько запрокинута назад, спина прямая, практически не сгибается и не двигается при ходьбе. При этом несколько избыточны движения в области тазового пояса, походка «разболтанная», шаг широкий, ноги ставит далеко друг от друга. Ретроколлис.
Речь правильная, с богатым словарным запасом, эмоционально окрашенная, с яркими интонациями. Темп речи умеренный, ближе к медленному, речь несколько неплавная, тягучая. Голос хорошо модулирует, однако по тембру «надтреснутый, шероховатый», имеет носовой оттенок. При проверке когнитивных функций с помощью краткого исследования когнитивного состояния, теста рисования часов, монреальской шкалы оценки когнитивных функций и батареи лобных тестов получены нормальные результаты.
Зрачки равные, округлой формы, обычного диаметра, живо реагируют на свет. Глазные щели D=S, движения глазных яблок по горизонтали в полном объеме, по вертикали – с особенностями: затруднены следящие движения вверх и вниз, трудности с фиксацией взора в этих положениях, при длительном нахождении неврологического молоточка в этих позициях больная очень быстро отводит взор. Болезненность, тошноту, головокружение, двоение в этих положениях глаз отрицает. Затруднены также движения глаз в верхние и нижние косые положения. Возможно, имеет место формирование пареза взора вверх и вниз. Корнеальные рефлексы живые, симметричные. Чувствительность на лице не изменена, нижнечелюстной рефлекс оживлен, симметричный. Лицо симметричное. Глотание сохранено. Язык по средней линии, без атрофий и фасцикуляций, розовый, влажный. Определяются умеренно выраженные симптомы орального автоматизма: хоботковый, назолабиальный. Объем активных движений в конечностях полный, парезов нет. Мышечный тонус повышен по экстрапирамидному типу во всех группах мышц, в том числе в аксиальной мускулатуре. Выявляется феномен восковой гибкости, феномен Нойка-Ганева ярко представлен с 2-х сторон. Глубокие рефлексы с рук умеренные, без разницы сторон, с ног – ближе к низким, также симметричные. Чувствительность в области конечностей, живота не изменена. От проверки чувствительности в аногенитальной зоне больная отказалась. В позе Ромберга больная легко пошатывается вправо. Пальце-носовую пробу выполняет с легкой интенцией справа. Пяточно-коленную пробу выполняет с атаксией справа. Выявляются нерезко выраженные адиадохокинез, гиперметрия, дисметрия слева. Менингеальных знаков нет.
Таким образом, по результатам расспроса пациентки и первичного осмотра были выявлены следующие нарушения: гипокинетико-ригидный синдром с преобладанием тонуса мышц в аксиальной мускулатуре и формированием разгибательной позиции тела, псевдобульбарный синдром, глазодвигательные нарушения, нарушения походки, постуральная неустойчивость и падения, мозжечковые расстройства, нарушения сна, вегетативные расстройства в виде нарушений мочеиспускания и артериальной гипертензии. Большинство этих симптомов могут встречаться при обоих обсуждаемых заболеваниях (синдром паркинсонизма, псевдобульбарный синдром, нарушения сна). Тем не менее, некоторые из них более характерны для МСА (мозжечковые симптомы, вегетативные расстройства), тогда как другие – для ПНП (разгибательная поза при ходьбе, постуральная неустойчивость, парезы взора по вертикали, особенно вниз).
Пациентке было рекомендовано дообследование:
- МРТ головного мозга.
- ЭЭГ.
- Осмотр окулиста, эндокринолога.
- Гликемический профиль, уровень гликозилированного гемоглобина, контроль электролитов (калий, натрий, кальций) в динамике.
- Измерение объема потребляемой жидкости за сутки и объема выделяемой мочи за сутки, общий анализ мочи, ведение дневника мочеиспусканий.
- Определение концентрации эстрогенов в крови, антидиуретического гормона.
Через 3 месяца пациентка была осмотрена повторно (в период госпитализации в урологическое отделение по поводу обострения хронического цистита). На ЭЭГ – вариант возрастной нормы, на МРТ головного мозга – легкое расширение боковых желудочков, на глазном дне – ангиопатия сетчаток, отклонений со стороны биохимических показателей, в содержании гормонов и электролитов выявлено не было, эндокринолог не выявил заболеваний эндокринной системы. Согласно дневнику мочеиспусканий, их количество колебалось в различные дни от 8 до 27 за сутки, ночных – от 1 до 7. За эти 3 месяца у пациентки появились жалобы на ухудшение памяти на текущие бытовые события, профессиональная память не страдает. Беспокоит изменение почерка: стал более небрежным, неаккуратным. За последние 3 месяца похудела на 2 кг. В плане нарушений мочеиспускания отмечает, что бывают «хорошие дни», когда учащенное мочеиспускание беспокоит мало, и «ужасные», когда больная постоянно бегает в туалет. Пациентка обратила внимание на следующее: приняв анальгетик для снятия головной боли, заметила снижение количества походов в туалет в этот вечер, в дальнейшем несколько раз «экспериментировала», принимая анальгетик в те дни, когда мочеиспускание было особенно частым, прием лекарства приводил к некоторому урежению мочеиспускания.
В неврологическом статусе определяются следующие изменения: повторное исследование когнитивных функций выявило наличие умеренных когнитивных нарушений: по краткому исследованию когнитивного состояния (MMSE) – 26 баллов, по Монреальской шкале (MoCA) – 24 балла. В наибольшей степени нарушилось отсроченное воспроизведение запоминаемых слов и счет. Другие тесты выполнялись хорошо. Отмечается парез взора вверх, затруднение движений глазных яблок вниз, нарушение плавности горизонтальных движений глаз. В остальном в неврологическом статусе динамики нет.
Принимая во внимание, что учащенное мочеиспускание в данный период может быть связано с обострением хронического цистита, а также тот факт, что сформировался парез взора вверх, ухудшились движения глазных яблок вниз, появились умеренные когнитивные расстройства в виде нарушений памяти и счета, более правомочным представлялся диагноз прогрессирующего надъядерного паралича, при котором отмечается синдром паркинсонизма с преобладанием тонуса в аксиальной мускулатуре и формированием разгибательной позиции, отсутствует на ранней стадии изменение длины шага, скорости ходьбы, типичны глазодвигательные нарушения, особенно вертикальные парезы взора или офтальмоплегия, а когнитивные нарушения, достаточно быстро достигающие степени деменции. Соответственно данному диагнозу, ожидалось прогрессирование симптомов паркинсонизма, усиление постуральной неустойчивости, глазодвигательных нарушений, дальнейшее ухудшение когнитивных функций с последующим развитием деменции.
Через 10 месяцев состоялся следующий осмотр больной, когда пациентка попала в неврологическое отделение больницы с острой нейропатией лицевого нерва справа на фоне переохлаждения. Пациентка отмечает, что эпизоды неустойчивости случаются с той же частотой, однако падений стало меньше, объясняет это тем, что теперь «внимательно смотрит под ноги». Основной жалобой остается учащенное мочеиспускание с императивными позывами. В неврологическом статусе помимо выраженной асимметрии половины лица, усилились нарушения почерка, появились черты мегалографии, в остальном динамики нет: сохраняется повышение мышечного тонуса во всех группах мышц по экстрапирамидному типу, в том числе в аксимальной мускулатуре, ретроколлис, однако выраженность его та же, ограничены движения глазных яблок вверх, объем движений вниз и по горизонтали достаточный, но движения неплавные. Сохраняются имевшиеся мозжечковые расстройства, когнитивные нарушения не прогрессируют: MMSE 27 баллов, MoCA – 24 балла, другие тесты – норма. Принимая во внимание тот факт, что типичного для ПНП быстрого неуклонного прогрессирования заболевания не отмечается, когнитивные функции остаются сохранными, пареза взора вниз, тем более полной офтальмоплегии – нет, отмечается нарастание мозжечковых расстройств, стойкость вегетативных нарушений в виде резко учащенного мочеиспускания с императивными позывами уже вне обострения хронического цистита, диагноз ПНП, согласно критериям NINDS-SPSP [7], требует пересмотра.
В данной ситуации более вероятным представляется диагноз мультисистемной атрофии, при которой также могут иметь место глазодвигательные нарушения, но они, как правило, не достигают степени пареза взора или офтальмоплегии, часто встречаются когнитивные нарушения, которые, однако, не достигают степени деменции. Что же касается разгибательной позиции туловища, то она является частым, но не облигатным симптомом ПНП.
Мнение о том, что у данной пациентки клиническая картина заболевания более соответствует диагнозу мультисистемной атрофии, чем ПНП, подтверждает следующее наблюдение: спустя 3,5 года от момента последней госпитализации в ВГКБСМП № 1, т.е. через 4 года и 7 месяцев от момента первого знакомства, с пациенткой состоялась случайная встреча в вестибюле больницы, куда она пришла проведать родственницу. Больная узнала врача, рассказала, что у нее усилились расстройства мочеиспускания, присоединились эпизоды неудержания мочи, постоянно пользуется памперсами, вынуждена была полгода назад оставить успешную юридическую практику, в настоящее время работает только как приглашенный консультант, а все остальное время заботится о сыне и помогает дочери с внуками: сопровождает в школу и на дополнительные занятия, проверяет уроки. Речь у больной несколько замедленная, с элементами дизартрии, эмоционально окрашенная, голос слегка охриплый, горизонтальные движения глаз сохранены (легко переводит взор с одного предмета на другой, движения вверх ограничены, вниз — немного), походка с явлениями мозжечковой атаксии, поза при ходьбе с запрокинутой головой, развернутыми назад плечами.
Заключение. Таким образом, на основании анализа представленного случая можно заключить, что клинический метод является ведущим в диагностике заболеваний, при которых отсутствуют патогномоничные симптомы, четко очерченные изменения при нейровизуализации, биохимические, инструментальные маркеры. Динамическое наблюдение за пациентами, у которых отмечаются симптомы, соответствующие нескольким заболеваниям со сходной клинической картиной, является крайне важным для уточнения диагноза. Как видно из описания данного случая, на более ранних этапах развития заболевания клиническая картина может больше соответствовать одному диагнозу, а на последующих – другому. В описанном наблюдении представляет интерес тот факт, что у пациентки сочеталось несколько симптомов, характерных для разных заболеваний, например, разгибательная позиция туловища и раннее развитие постуральной неустойчивости, более характерные для ПНП, и вегетативные нарушения и мозжечковые симптомы, более характерные для МСА. Возможно, это связано с некоторыми элементами патогенеза, общими для двух заболеваний [8].
Пнп и пкп неврология — Медицина мира
При поражении мозжечка характерны расстройства статики и координации движений, мышечная гипотония и нистагм. Поражение мозжечка, прежде всего его червя, ведет к нарушениям статики — возможности поддержания стабильного положения центра тяжести тела человека, равновесия, устойчивости. При расстройстве указанной функции возникает статическая атаксия (от греч. ataxia — беспорядок, неустойчивость). Отмечается неустойчивость больного. Поэтому в положении стоя он широко расставляет ноги, балансирует руками. Особенно четко статическая атаксия выявляется при искусственном уменьшении площади опоры, в частности в позе Ромберга. Больному предлагается встать, плотно сдвинув ступни и слегка приподняв голову. При наличии мозжечковых расстройств отмечается неустойчивость больного в этой позе, тело его раскачивается, иногда его «тянет» в какую-то определенную сторону, при этом, если больного не поддержать, он может упасть. В случае поражения червя мозжечка больной обычно раскачивается из стороны в сторону и чаще падает назад. При патологии полушария мозжечка возникает тенденция к падению преимущественно в сторону патологического очага. Если расстройство статики выражено умеренно, его легче выявить в так называемой усложненной или сенсибилизированной позе Ромберга. Больному предлагается поставить ступни на одну линию, чтобы носок одной ступни упирался в пятку другой. Оценка устойчивости та же, что и в обычной позе Ромберга. В норме, когда человек стоит, мышцы его ног напряжены {реакция опоры), при угрозе падения в сторону нога его на этой стороне перемещается в том же направлении, а другая нога отрывается от пола {реакция прыжка). При поражении мозжечка (главным образом червя) у больного нарушаются реакции опоры и прыжка. Нарушение реакции опоры проявляется неустойчивостью больного в положении стоя, особенно в позе Ромберга. Нарушение реакции прыжка приводит к тому, что если врач, встав позади больного и подстраховывая его, толкает больного в ту или иную сторону, то больной падает при небольшом толчке (симптом толкания). При поражении мозжечка походка больного обычно изменена в связи с развитием статолокомоторной атаксии. «Мозжечковая» походка во многом напоминает походку пьяного человека, поэтому се иногда называют «походкой пьяного». Больной из-за неустойчивости идет неуверенно, широко расставляя ноги, при этом его «бросает» из стороны в сторону. А при поражении полушария мозжечка он отклоняется при ходьбе от заданного направления в сторону патологического очага. Особенно отчетлива неустойчивость при поворотах. Если атаксия оказывается резко выраженной, то больные полностью теряют способность владеть своим телом и не могут не только стоять и ходить, но даже сидеть. Преимущественное поражение полушарий мозжечка ведет к расстройству его противоинерционных влияний, в частности к возникновению кинетической атаксии. Она проявляется неловкостью движений и особенно выражена при движениях, требующих точности. Для выявления кинетической атаксии проводятся пробы на координацию движений. Далее приводится описание некоторых из них. Проба на диадохокинез (от греч. diadochos — последовательность). Больному предлагается закрыть глаза, вытянуть вперед руки и быстро, ритмично супи-нировать и пронировать кисти рук. В случае поражения полушария мозжечка движения кисти на стороне патологического процесса оказываются более размашистыми (следствие дисметрии, точнее — гиперметрии), в результате кисть начинает отставать. Это свидетельствует о наличии адиадохокинеза. Пальценосовая проба. Больной с закрытыми глазами должен отвести руку, а затем, не торопясь, указательным пальцем дотронуться до кончика носа. В случае мозжечковой патологии рука на стороне патологического очага совершает избыточное по объему движение (гиперметрия), в результате чего больной промахивается. При пальценосовой пробе выявляется характерный для мозжечковой патологии мозжечковый (интенционный) тремор, амплитуда которого нарастает по мере приближения пальца к цели. Эта проба позволяет выявить и так называемую брадителекинезию (симптом узды): недалеко от цели движение пальца замедляется, иногда даже приостанавливается, а затем возобновляется вновь. Пальце-пальцевая проба. Больному с закрытыми глазами предлагается широко развести руки и затем сближать указательные пальцы, стремясь попасть пальцем в палец, при этом, как и при пальценосовой пробе, выявляются ин-тенционное дрожание и симптом узды. Пятонно-коленная проба (рис. 7.3). Больному, лежащему на спине с закрытыми глазами, предлагают высоко поднять одну ногу и затем ее пяткой попасть в колено другой ноги. При мозжечковой патологии больной не может или ему трудно попасть пяткой в колено другой ноги, особенно выполняя пробу ногой, гомолатеральной пораженному полушарию мозжечка. Если все-таки пятка достигает колена, то предлагается провести ею, слегка касаясь передней поверхности голени, вниз, к голеностопному суставу, при этом в случае мозжечковой патологии пятка все время соскальзывает с голени то в одну, то в другую сторону. Рис. 7.3. Пяточно-коленная проба. Указательная проба. Больному предлагается несколько раз указательным пальцем попасть в резиновый наконечник молоточка, находящегося в руке обследующего. В случае мозжечковой патологии в руке пациента на стороне пораженного полушария мозжечка отмечается мимопопадание вследствие дис-метрии. Симптом Тома—Жюменти. Если пациент берет предмет, например стакан, он при этом чрезмерно раздвигает пальцы. Мозжечковый нистагм. Подергивание глазных яблок при взгляде в стороны (горизонтальный нистагм) рассматривается как следствие интенционного дрожания глазных яблок (см. главу 30). Расстройство речи. Речь теряет плавность, становится взрывчатой, фраг-ментированной, скандированной по типу мозжечковой дизартрии (см. главу 25). Изменение почерка. В связи с расстройством координации движений руки почерк становится неровным, буквы деформированы, чрезмерно крупные (ме-галография). Пронаторный феномен. Больному предлагается удерживать вытянутые вперед руки в положении супинации, при этом на стороне пораженного полушария мозжечка вскоре происходит спонтанная пронация. Симптом Гоффа—Шильдера. Если больной держит руки вытянутыми вперед, то на стороне пораженного полушария рука вскоре отводится кнаружи. Имитационный феномен. Больной с закрытыми глазами должен быстро придать руке положение, аналогичное тому, которое обследующий перед этим придал другой его руке. При поражении полушария мозжечка гомолатеральная ему рука совершает движение, избыточное по амплитуде. Феномен Дойникова. Пальцевой феномен. Сидящему пациенту предлагается супинированные кисти с разведенными пальцами положить на свои бедра и закрыть глаза. В случае поражения мозжечка на стороне патологического очага вскоре возникает спонтанное сгибание пальцев и пронация кисти и предплечья. Симптом Стюарта—Холмса. Исследующий просит сидящего на стуле пациента сгибать супинированные предплечья и в то же время, взяв его руки за запястья, оказывает ему сопротивление. Если при этом неожиданно отпустить руки пациента, то рука на стороне поражения, сгибаясь по инерции, с силой ударит его в грудь. Гипотония мышц. Поражение червя мозжечка ведет обычно к диффузной мышечной гипотонии. При поражении полушария мозжечка пассивные движения выявляют снижение мышечного тонуса на стороне патологического процесса. Гипотония мышц ведет к возможности переразгибания предплечья и голени (симптом Ольшанского) при пассивных движениях, к появлению симптомов «болтающейся» кисти или стопы при их пассивном встряхивании. Патологические мозжечковые асинергии. Нарушения физиологических синергии при сложных двигательных актах выявляются, в частности, при следующих пробах (рис. 7.4). 1. Асинергия по Бабинскому в положении стоя. Если стоящий со сдвинутыми ногами пациент пытается прогнуться назад, запрокинув при этом голову, то в норме в таком случае происходит сгибание коленных суставов. При мозжечковой патологии в связи с асинергией это содружественное движение отсутствует, и больной, теряя равновесие, падает назад. Рис. 7.4. Мозжечковая асинергия. 1 — походка больного с выраженной мозжечковой атаксией; 2 — наклон туловиша назад в норме; 3 — при поражении мозжечка больной, наклоняясь назад, не может сохранить равновесия; 4 — выполнение пробы на мозжечковую асинергию по Бабинскому здоровым человеком; 5 — выполнение той же пробы больным с поражением мозжечка. 2. Асинергия по Бабинскому в положении дежа. Больному, лежащему на твердой плоскости с вытянутыми ногами, разведенными на ширину надплечий, предлагается скрестить руки на груди и затем сесть. При наличии мозжечковой патологии в связи с отсутствием содружественного сокращения ягодичных мышц (проявление асинергии) больной не может фиксировать на площади опоры ноги и таз, в результате ноги поднимаются и сесть ему не удается. Не следует переоценивать значимость этого симптома у пожилых пациентов, у людей с дряблой или ожиревшей брюшной стенкой. Резюмируя изложенное, следует подчеркнуть многообразие и важность выполняемых мозжечком функций. Являясь частью комплексного регуляторно-го механизма с обратной связью, мозжечок выполняет роль координационного центра, обеспечивающего равновесие тела и поддержание мышечного тонуса. Как отмечает P. Duus (1995), мозжечок обеспечивает возможность выполнения дискретных и точных движений, при этом автор обоснованно считает, что мозжечок работает подобно компьютеру, отслеживая и координируя сенсорную информацию на входе и моделируя моторные сигналы на выходе.
Читать далее
Источник: www.myneuro.ru
Читайте также
Ударим волной по спине больной!
Мы живем в эпоху сидячего образа жизни. «Благодаря» ему каждый из нас рано или поздно сталкивается с болями в спине и суставах.
Лечение опорно-двигательного аппарата
Для лечения опорно-двигательного аппарата в клинике «МЕДИК» есть свой новейший аппарат, дающий наилучший эффект. Это аппарат ударно-волновой терапии (УВТ) Duolith SD1 (Швейцария) — создан мировым лидером в производстве ударно-волновых систем — компанией Storz Medical. Аппараты для ударно-волновойтерапии от Storz Medical – это гарантия высоких технологических стандартов. Все компоненты систем подвергаются серьезным испытаниям и тестированиям, что обеспечивает максимальную эффективность, функциональность и надежность каждого аппарата.
Ударно-волновая терапия (УВТ)
DUOLITH SD1 успешно применяется для междисциплинарной терапии:
- Ортопедия и хирургия: большие плечевые кальцификаты, пяточная боль, триггерные точки, тендинит зон прикрепления, акупунктура без игл, псевдоартрозы
- Неврология: улучшение подвижности после инсульта, посттравматического спазма, полиневропатия (PNP)
- Урология: хронический тазовый болевой синдром (CPPS), болезнь Пейрони (penile curvature), терапия триггерных точек
- Косметология: целлюлит, дряблость кожи, растяжки, рубцы, шрамы
В России УВТ начали активно использовать после 2000-го года, переняв этот способ у швейцарских специалистов. Новая методика очень быстро зарекомендовала себя с хорошей стороны. Суть метода УВТ заключается в том, что акустические волны инфразвуковой частоты, которые создает специальный аппарат, воздействуют на пораженные ткани, разрушая микроскопические кристаллы кальция, которые нарушают правильное строение и вызывают боль в суставах, заметно улучшая кровообращение и активизируя восстановительные процессы.
Результаты после ударно-волновой терапии
В результате улучшается метаболизм в тканях, проходит боль, исчезают отеки. В сущности, ударные волны – это просто звук, но его частота ниже порога человеческого восприятия. Электрический импульс, который создает аппарат, преобразуется в звуковую волну. Она проникает в ткани и поглощается, достигнув кости.
УВТ помогает справится со многими заболеваниями, такими как: остеохондроз, артроз, хронические боли в суставах рук и ног, пяточная «шпора», грыжи межпозвоночных дисков и другие заболевания костей и суставов. Кроме того, она весьма эффективна при восстановлении после травм и переломов. Использование УВТ позволяет многим людям с патологическими деформациями суставов остановить дальнейшее развитие артроза и избежать хирургического вмешательства.
Как проводится процедура УВТ в клинике «МЕДИК»?
Процедура проведения УВТ очень проста и безболезненна. Пациент удобно располагается на кушетке, а врач покрывает тонким слоем специального геля те участки кожи, которые будут соприкасаться с датчиком аппарата. Затем врач задает индивидуальные настройки аппарата, которые зависят от каждого конкретного случая и патологии, которую будут лечить. Датчик плотно прижимается к коже и процесс начинается. Процедура длится от 5 до 30 минут. Сеансы повторяют в среднем через каждые 7-10 дней, а полный курс лечения состоит из 5-7 процедур. Однако большинство пациентов отмечает, что боль становится меньше после первой же процедуры.
Назначает УВТ физиотерапевт клиники «МЕДИК». Для проведения сеанса не требуется никакой специальной подготовки, а после него нет необходимости в реабилитации. К тому же этот метод, в отличие от медикаментозного лечения, не имеет побочных эффектов.
Ударно-волновая терапия в Чебоксарах
Провести процедуру УВТ можно в нашей клинике «МЕДИК»! Записывайтесь по телефону 8 (8352) 23-77-23
| |||||
| ✎ Создать тему | Личное сообщение | Имя | Дата | |||
| 20 | 295 | «I recall object V-ing …», grammar rule | VictorM2 | 11.08.2021 | 10:59 |
| 1 | 24 | Happy Birthday 2 Lonely Knight | Себастьян Перейра, торговец… | 13.08.2021 | 0:01 |
| 16 | 359 | Перевод «застой в мышцах» на английский и другие языки, раз уж на то пошло | Chia | 10.08.2021 | 20:16 |
| 62 | 3790 | ОФФ: А давайте поговорим о просмотренных сериалах или фильмах? | 1 2 3 все | qp | 28.06.2021 | 1:15 |
| 272 | 8372 | Предложения и вопросы по работе нового сайта | 1 2 3 4 5 6 7 все | 4uzhoj | 15.05.2019 | 11:02 |
| 2 | 119 | «Горячий» пласт | Розмари | 12.08.2021 | 8:19 |
| 2 | 46 | Discontinued profile | A111981 | 12.08.2021 | 20:32 |
| 481 | 8410 | Ошибки в словаре | 1 2 3 4 5 6 7 8 9 10 11 все | 4uzhoj | 23.02.2021 | 13:36 |
| 4 | 356 | Power of attorney/Сompany Referral | Valentinochka | 5.08.2021 | 6:55 |
| 6 | 185 | В рамках дисциплины | Tae_tae | 11.08.2021 | 20:30 |
| 31 | 479 | Мемсорс | wise crocodile | 10.08.2021 | 9:53 |
| 7 | 307 | lever | littlemoor | 10.08.2021 | 0:38 |
| 13 | 470 | Within 30 days Due net | Aniss | 28.07.2021 | 17:35 |
| 9 | 382 | load failure | VictorMashkovtsev | 9.08.2021 | 10:28 |
| 5 | 370 | Перевод времени в российских официальных документах | astrmarina | 10.08.2021 | 7:16 |
| 6 | 362 | Согласно справке из реестра | Alex16 | 7.08.2021 | 21:44 |
| 12 | 607 | Прикинь | Jill_P | 6.08.2021 | 13:54 |
| 2 | 283 | Settlement Drum | vellendorf | 8.08.2021 | 8:47 |
| 6 | 638 | OFF: БП Effectiff — какие отзывы? | Xtkjdtr | 6.08.2021 | 19:13 |
| 57 | 2546 | Журнал для технических переводчиков «Petrotran» | 1 2 3 все | niccolo | 22.07.2021 | 15:45 |
| 2 | 374 | electrolyzers E/F | OZ_MaLL | 6.08.2021 | 8:43 |
| 188 | 5847 | Переворот рынка перевода — выход китайского на 1 место по востебованности | 1 2 3 4 5 6 7 8 все | niccolo | 2.08.2021 | 13:14 |
| 10 | 454 | Как сказать на англ «выпускник специальности» | Tae_tae | 6.08.2021 | 18:12 |
отчет о первом случае из Индии
BMJ Case Rep. 2011; 2011: bcr0920114804.
Редкое заболевание
Маниша Раджан Мадкаикар
1 Отдел детской иммунологии и биологии лейкоцитов, Национальный институт иммуногематологии, Мумбаи, Индия
Шилпа Кулкарни
2 Больница для детей в Джербаи, Мумбай , Индия
Prashant Utage
2 Отделение неврологии, Детская больница Бай Джербай Вадиа, Мумбаи, Индия
Линетт Фэрбенкс
3 Исследовательская лаборатория пуринов, Больница Святого Томаса, Лондон, Великобритания
Канджакша
4 Национальный институт иммуногематологии, Мумбаи, Индия
Энтони Маринаки
3 Исследовательская лаборатория пуринов, больница Св. Томаса, Лондон, Великобритания
Мукеш Десаи
2 Отделение неврологии Джербаи, Байбай, Байрология Больница для детей, Мумбаи, Индия
1 Pediatric Immuno logy и отделение биологии лейкоцитов, Национальный институт иммуногематологии, Мумбаи, Индия
2 Отделение неврологии, Детская больница Бай Джербай Вадиа, Мумбаи, Индия
3 Исследовательская лаборатория пуринов, Больница Святого Томаса, Лондон, Великобритания
4 Национальный институт иммуногематологии, Мумбаи, Индия
Эта статья цитируется в других статьях в PMC.Abstract
Авторы впервые сообщают о случае дефицита пуриновой нуклеозидфосфорилазы (PNP) из Индии. Случай представил рецидивирующие тяжелые инфекции, задержку развития, судороги и прогрессирующее неврологическое ухудшение. Несмотря на рецидив инфекции, диагноз первичного иммунодефицита был отложен из-за преобладающих неврологических симптомов. Секвенирование гена PNP выявило новую мутацию, приводящую к преждевременному стоп-кодону.
Предпосылки
Пуриновая нуклеозидфосфорилаза (PNP) — это широко распространенный фермент, необходимый для разложения пуриновых нуклеозидов. 1 PNP экспрессируется на высоких уровнях в тимусе и лимфатических узлах, где он удаляет дезоксигуанозин, образующийся в результате распада ДНК. PNP расщепляет пурин-сахарную связь гуанозина, инозина, дезоксигуанозина и дезоксиинозина, высвобождая гуанин и гипоксантин. Эти основания либо используются в качестве предшественников АТФ и гуанозинтрифосфата (ГТФ), либо окисляются до мочевой кислоты. В отсутствие PNP его нуклеозидные субстраты становятся повышенными, и продукция мочевой кислоты снижается. 2
Кроме того, дезоксигуанозин аномально фосфорилируется дезоксицитидинкиназой, ядерным ферментом, или дезоксигуанозинкиназой, митохондриальным ферментом.Результирующая экспансия дезоксигуанозинтрифосфата (dGTP) подавляет синтез и восстановление ДНК. Эти вредные эффекты dGTP наиболее очевидны в лимфоидной ткани, подвергающейся быстрому обновлению клеток, особенно в Т-лимфоцитах, подвергающихся созреванию тимуса. Неврологические нарушения при дефиците PNP могут быть результатом нарушения функции митохондрий или истощения ГТФ в нейронах. 2 3
Дефицит PNP (OMIM 164050) — редкая форма тяжелого комбинированного иммунодефицитного расстройства (SCID) с аутосомно-рецессивным наследованием.Это приводит к глубокому дефекту Т-клеток с вариабельной дисфункцией В-клеток 4 и впервые было зарегистрировано в 1975 году у ребенка с рецидивирующей инфекцией и анемией. 5 Пациенты с недостаточностью PNP обычно обращаются с нарушением нормального развития, рецидивирующими и тяжелыми инфекциями, неврологической дисфункцией и аутоиммунитетом.
Дефицит PNP составляет только 4% пациентов с ТКИН. Было зарегистрировано менее 50 случаев дефицита PNP и, насколько нам известно, ни одного из Индии. Мы сообщаем о клинических и иммунологических результатах исследования индийской девочки с дефицитом PNP и молекулярного дефекта, ответственного за него.
Описание клинического случая
Четвертый брат и сестра, которому исполнилось 22 месяца, родившаяся от кровного брака третьей степени, была направлена к нам с рецидивирующими инфекциями нижних дыхательных путей, недостаточным развитием и глобальной неврологической дисфункцией. Она впервые обратилась на 15-й день жизни с пневмонитом и кандидозом полости рта. Ей трижды требовалась госпитализация по аналогичным жалобам в первый год. У нее также были проблемы с ростом и глобальные задержки в развитии с рождения. Однако ребенок каждый раз получал симптоматическое лечение без каких-либо иммунологических исследований.В 12 месяцев ребенок поступил с пневмонией с поражением заднего сегмента верхней доли справа с респираторным дистрессом, а также с замеченными непроизвольными движениями. Она также получила эмпирическую противотуберкулезную терапию по поводу инфекций грудной клетки и неврологических проблем. Наконец, в возрасте 22 месяцев ее направили в отделение иммунологии, чтобы исключить основное заболевание иммунодефицита. При обследовании пациентка плохо питалась, показатели роста были ниже третьего процентиля.Она была иммунизирована ОПВ-1, БЦЖ, но рубца от БЦЖ не было. У нее был оральный молочница. При обследовании грудной клетки выявлены двухсторонние базальные крепитации. Неврологическое обследование выявило общую задержку развития. У нее была аксиальная гипотония, повышенный тонус в обеих нижних конечностях с живыми глубокими сухожильными рефлексами.
Исследования
Исследования показали Hg 9,2 г%, количество лейкоцитов 18 200 / мм 3 с абсолютным количеством лимфоцитов 364 / мм 3 ) и количество тромбоцитов 546 000 / мм 3 .В связи со значительными неврологическими проблемами пациентки, она была дополнительно обследована с помощью исследований спинномозговой жидкости (ЦСЖ) и МРТ головного мозга. ЦСЖ выявила картину, указывающую на менингит, с 14 клетками / мм 3 (11% нейтрофилов и 89% лимфоцитов, глюкоза 44 мг%, белки 21 мг%). Посев ЦСЖ и повторные посевы мокроты не привели к росту каких-либо организмов. МРТ показала усиление базального лептоменингиального отдела в супратентальной области сзади, что свидетельствует о воспалительном процессе, таком как менингит.Иммунологическое исследование выявило лимфопению с низким фенотипом Т, В и естественных киллерных клеток. Ввиду неврологических симптомов, уровень мочевой кислоты в сыворотке был низким (1 мг%) (нормальный диапазон 2–7 мг%). Активность ПНФ в лизате эритроцитов у пациентки не определялась, тогда как ее родители и ее брат демонстрировали активность диапазона носителей (мать: 1328 нмоль / час / мг Hb, отец: 1786 нмоль / час / мг Hb, брат: 2040 нмоль / час / мг Hb с нормальным диапазон регулирования 3000–7000 нмоль / ч / мг Hb). У ее сестры был нормальный уровень PNP.
Таблица 1
Иммунологические исследования
| Пациент | Нормальный диапазон | ||||
|---|---|---|---|---|---|
| (лимфоциты) оценка подмножеств / мм28 | |||||
| CD4 | 109 | 1400–4300 | |||
| CD8 | 7 | 500–1700 | |||
| CD19 | 153 | 61012–2600 | 160–950 | ||
| Уровни иммуноглобулинов г / л | IgG | 8.19 | 4,0–15,9 | ||
| IgA | 1,75 | 0,01–0,91 | |||
| IgM | 2,17 | 0,34–2,06 |
Секвенирование кодирующей области гена PNP и фланкирующих интронных последовательностей выявило у пациента новую гомозиготную несмысловую мутацию c.199C> T, PNP p.67R> X. Эта мутация приводит к преждевременному стоп-кодону в экзоне 3, предсказывая укороченный белок. Родители были гетерозиготными по мутации.
Дифференциальный диагноз
Другие формы ТКИН с Т-В-фенотипом, такие как недостаточность аденозиндеманиназы (АДА), остаются основным дифференциальным диагнозом. Однако уровни ADA у ребенка были в пределах нормы.
Лечение
Ребенок получил симптоматическое лечение антибиотиками и противогрибковыми препаратами.
Исход и наблюдение
У пациента развилось прогрессирующее неврологическое ухудшение с тяжелыми дистоническими движениями, и повторная МРТ выявила атрофию коры головного мозга и мозжечка и потерю миелинизации белого вещества. Также наблюдалось сокращение хвостатых ядер. В конце концов, пациент умер от сепсиса.
Обсуждение
Дефицит PNP приводит к дефекту Т-клеток и, таким образом, делает пациентов восприимчивыми к различным опасным для жизни инфекциям, вызываемым обычными условно-патогенными микроорганизмами, такими как Candida albicans и Pneumocystis jiroveci .Распространенная ветряная оспа, стойкие инфекции, вызванные вирусом простого герпеса, очень тяжелы или даже в некоторых случаях приводят к летальному исходу. Это также связано с повышенным риском автоматических заболеваний. Однако эти симптомы разнообразны и могут не проявляться в первые несколько месяцев жизни. 1 — 6
Неврологические проблемы, включая задержку развития, гипертонию, спастичность, тремор, атаксию, задержку двигательного развития, поведенческие трудности и умственную отсталость различной степени, наблюдаются более чем у половины пациентов. 1 Таким образом, пациенты с неврологическими нарушениями и рецидивирующими инфекциями должны быть тщательно обследованы на иммунодефицит. Пациентов с лимфопенией и низким абсолютным количеством Т-лимфоцитов также следует рассматривать на предмет дефицита PNP. Низкий уровень мочевой кислоты является признаком дефицита PNP. Следовательно, скрининг уровня мочевой кислоты у пациентов с лимфопенией может позволить более раннюю диагностику дефицита PNP, даже до того, как синдром полностью проявится. Неопределяемый уровень активности фермента PNP в лизатах эритроцитов подтверждает диагноз PNP. 7 Молекулярная характеристика дефицита PNP помогает в подтверждении диагноза, генетическом консультировании и пренатальной диагностике. У пациентов с дефицитом PNP описано около 40 различных мутаций. Однако мутация c.199C> T (p.67R> X), обнаруженная у этого пациента, никогда ранее не описывалась. Эта мутация привела к тяжелому дефициту PNP с неопределяемой каталитической активностью.
Прогноз недостаточности PNP при отсутствии лечения плохой, так как большинство пациентов умирают от тяжелых инфекций.Единственное доступное лекарство от дефицита PNP — трансплантация гемопоэтических стволовых клеток (HSCT). 8 Хотя HSCT восстанавливает метаболизм пуриновых нуклеозидов в популяциях ненейрональных клеток, отчеты предполагают, что он также может стабилизировать нейрональную дисфункцию у этих пациентов. 9
Очки обучения
- ▶ Широко доступные простые исследования, такие как иммунофенотипирование лимфоцитов, определение уровня иммуноглобулина и определение мочевой кислоты в сыворотке, могут дать очень важные подсказки для диагностики дефицита PNP.
- ▶ Неврологические симптомы могут отвлекать внимание от исследования и в некоторых случаях задерживать постановку диагноза.
- ▶ В этом случае была обнаружена новая мутация в гене PNP.
Сноски
Конкурирующие интересы Нет.
Согласие пациента Получено.
Ссылки
1. Markert ML. Дефицит пуриновой нуклеозидфосфорилазы. Immunodefic Rev 1991; 3: 45–81 [PubMed] [Google Scholar] 2. Хершфилд М, Митчелл Б.С. Иммунодефицитные заболевания, вызванные дефицитом аденозиндезаминазы и пуриновой нуклеозидфосфорилазы.В: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. Метаболические и молекулярные основы наследственных заболеваний. Восьмое издание Нью-Йорк: Макгроу-Хилл; 2001: 2585–625 [Google Scholar] 3. Симмондс Х.А., Фэрбенкс Л.Д., Моррис Г.С. и др. Дисфункция центральной нервной системы и истощение гуанозинтрифосфата эритроцитов при дефиците пуриновой нуклеозидфосфорилазы. Arch Dis Child 1987; 62: 385–91 [Бесплатная статья PMC] [PubMed] [Google Scholar] 4. Осборн WR, Ochs HD. Иммунодефицитное заболевание из-за дефицита пуриновой нуклеозидфосфорилазы.В: Ochs HD, Smith CIE, Puck JM, eds. Первичные иммунодефицитные заболевания. Молекулярно-генетический подход. Нью-Йорк: Издательство Оксфордского университета; 1999: 140–5 [Google Scholar] 5. Гиблетт Э. Р., Амманн А. Дж., Вара Д. В. и др. Дефицит нуклеозид-фосфорилазы у ребенка с сильно нарушенным Т-клеточным иммунитетом и нормальным В-клеточным иммунитетом. Lancet 1975; 1: 1010–3 [PubMed] [Google Scholar] 6. Сасаки Ю., Исэки М., Ямагути С. и др. Прямое доказательство аутосомно-рецессивного наследования Arg24 на терминирующий кодон в гене пуриновой нуклеозидфосфорилазы в семье с пациентом с тяжелым комбинированным иммунодефицитом.Hum Genet 1998; 103: 81–5 [PubMed] [Google Scholar] 7. Коэн А., Гудас Л. Дж., Амманн А. Дж. И др. Дезоксигуанозинтрифосфат как возможный токсический метаболит при иммунодефиците, связанном с дефицитом пуриновой нуклеозидфосфорилазы. J Clin Invest 1978; 61: 1405–9 [Бесплатная статья PMC] [PubMed] [Google Scholar] 8. Broome CB, Graham ML, Saulsbury FT и др. Коррекция дефицита пуриновой нуклеозидфосфорилазы путем трансплантации аллогенного костного мозга от брата или сестры. J Pediatr 1996; 128: 373–6 [PubMed] [Google Scholar] 9.Майерс Л.А., Хершфилд М.С., Нил В.Т. и др. Дефицит пуриновой нуклеозидфосфорилазы (PNP-def), проявляющийся лимфопенией и задержкой развития: успешная коррекция с помощью трансплантации пуповинной крови. J Pediatr 2004; 145: 710–2 [PubMed] [Google Scholar]Дефицит пуриновой нуклеозидфосфорилазы: MedlinePlus Genetics
Дефицит пуриновой нуклеозидфосфорилазы — это нарушение иммунной системы, называемое иммунодефицитом. Иммунодефицитные состояния — это состояния, при которых иммунная система не может эффективно защищать организм от чужеродных захватчиков, таких как бактерии и вирусы.
Люди с дефицитом пуриновой нуклеозидфосфорилазы имеют небольшое количество клеток иммунной системы, называемых Т-лимфоцитами, которые обычно распознают и атакуют чужеродных захватчиков, чтобы предотвратить заражение. У некоторых пораженных людей также есть небольшое количество других клеток иммунной системы, называемых В-клетками, которые обычно помогают бороться с инфекциями, производя иммунные белки, называемые антителами (или иммуноглобулинами). Эти белки нацелены на чужеродных захватчиков и помечают их для разрушения. Наиболее серьезно пораженные люди, у которых отсутствуют Т-клетки и В-клетки, имеют серьезное состояние, называемое тяжелым комбинированным иммунодефицитом (ТКИД).
Нехватка клеток иммунной системы у людей с дефицитом пуриновой нуклеозидфосфорилазы приводит к повторяющимся и стойким инфекциям, обычно начинающимся в младенчестве или раннем детстве. Инфекции чаще всего поражают носовые пазухи и легкие. Эти инфекции часто вызываются «условно-патогенными» организмами, которые обычно не вызывают заболеваний у людей с нормальной иммунной системой. Инфекции могут быть очень серьезными или опасными для жизни, и без успешного лечения для восстановления иммунной функции дети с дефицитом пуриновой нуклеозидфосфорилазы обычно не доживают до детства.
Младенцы с дефицитом пуриновой нуклеозидфосфорилазы обычно растут медленнее, чем здоровые дети. Около двух третей людей с этим заболеванием также имеют неврологические проблемы, которые могут включать задержку в развитии, умственную отсталость, трудности с равновесием и координацией (атаксия) и ригидность мышц (спастичность). Люди с дефицитом пуриновой нуклеозидфосфорилазы также подвержены повышенному риску развития аутоиммунных заболеваний, которые возникают, когда иммунная система нарушает работу и атакует ткани и органы организма.
Дефицит пуринуклеозидфосфорилазы — обзор
vb) Дефицит PNP.
Дефицит PNP также может приводить к SCID, потому что этот гомотримерный фермент катализирует стадию катаболизма пуринов после стадии ADA (см. Фиг. 24-3). PNP действует на инозин и дезоксиинозин, а также на гуанозин и дезоксигуанозин, высвобождая основные основные структуры гипоксантина и гуанина, соответственно. Эти молекулы, в свою очередь, являются субстратами для ксантиноксидазы и гуаниноксидазы соответственно, которые превращают их в ксантин.Затем ксантиноксидаза превращает ксантин в мочевую кислоту, которая попадает в кровь и выводится из организма. PNP обычно присутствует во всех клетках, но наиболее высоко экспрессируется в эритроцитах, гранулоцитах, периферических лимфоцитах и клетках почек. Пренатальная диагностика может быть проведена путем анализа активности PNP в культивируемых ворсинах хориона или амниотических клетках.
Поскольку для выживания достаточно 1,5% нормальной активности PNP (измеренной в эритроцитах), PNP SCID встречается гораздо реже, чем ADA SCID, и составляет только 4% всех случаев SCID.Глубокий дефицит PNP приводит к летальному исходу из-за обширных инфекций. У пациентов, гомозиготных по мутации PNP, функция Т-клеток может быть нормальной при рождении, что объясняет практически нормальные гуморальные реакции, наблюдаемые у очень молодых пациентов с PNP. Однако по мере роста пациента функция Т-клеток снижается или может колебаться между нормальной и нарушенной, что отрицательно влияет на ответы В-клеток. Пациенты с ПНП часто впервые попадают в поле зрения врачей из-за инфекций ветряной оспы, которые широко распространены из-за отсутствия функционирующих Т-клеток.Часто встречаются рецидивирующие инфекции пазух, мочевыводящих путей и среднего уха, а также диарея и пневмония. Гипоурикемия (снижение уровня уратов в крови) также может присутствовать из-за блокировки производства субстратов для ксантиноксидазы. Также часто встречаются дефекты ЦНС, приводящие к аномалиям скелета, влияющим на двигательную функцию. Аутоиммунитет встречается чаще при PNP SCID, чем при ADA SCID, поражая 30% пациентов с PNP SCID.
Иммунодефицит возникает при дефиците PNP, потому что, как и dATP, dGTP может ингибировать рибонуклеотидредуктазу и репликацию ДНК.В отсутствие процессинга пуриновых нуклеотидов с помощью PNP, dGTP накапливается в клетках и отравляет их, что приводит к лимфоцитопении. Однако повреждение dGTP происходит только в T- и NK-клетках, но не в B-клетках. Из-за внутренних различий в активности нуклеотидкиназ в предшественниках T / NK и B лимфоцитов, dGTP накапливается до высоких уровней в клетках линии T / NK, но не в клетках линии B. В результате количество В-клеток у этих пациентов нормальное.
Мыши с химически индуцированными точечными мутациями PNP демонстрируют фенотип, сходный с фенотипом PNP-дефицитных людей и хорошо коррелирующий с величиной остаточной активности PNP, сохраняющейся у мутантного животного.Было обнаружено, что накопление dGTP происходит преимущественно в митохондриях Т-клеток, что приводит к дефектам репарации митохондриальной ДНК. Предполагается, что это отсутствие репарации делает мышиные PNP-дефицитные Т-клетки сверхчувствительными к спонтанному повреждению митохондриальной ДНК. Это повреждение заставляет Т-клетки подвергаться массовому апоптозу, что приводит к лимфоцитопении.
Замена фермента с использованием формы очищенного PNP, конъюгированной с ПЭГ, может использоваться для лечения дефицита PNP у человека. Циркулирующий PNP-PEG обрабатывает любой инозин, дезоксиинозин, гуанозин и дезоксигуанозин, высвобождаемые из клеток в кровь, снижая вероятность поглощения лимфоцитами.Как и в случае с ADA-PEG, этот метод терапии безопасен и эффективен, но очень дорог. Интерес к максимальной стабильности инъекционного PNP привел к исследованию гексамерного PNP Escherichia coli в качестве субстрата для конъюгации с PEG. Бактериальный фермент PNP более стабилен при 37 °, чем ферменты PNP человека, мыши или крупного рогатого скота, а его гексамерная структура позволяет конъюгировать большее количество молекул PEG. В испытаниях лечения на мышах бактериальный PNP-PEG показал пониженную иммуногенность, но увеличенный период полужизни в крови и такую же ферментативную активность, как и мышиный PNP-PEG.Генная терапия также была предпринята при дефиците PNP, но только на экспериментальном уровне (см. Вставку 24-2).
Дефицит пуриннуклеозидфосфорилазы: основы практики, предпосылки, патофизиология
la Marca G, Giocaliere E, Malvagia S, Villanelli F, Funghini S, Ombrone D, et al. Разработка и валидация теста 2-го уровня для идентификации пациентов с дефицитом пуриновой нуклеозидфосфорилазы во время расширенного скрининга новорожденных с помощью жидкостной хроматографии и тандемной масс-спектрометрии. Clin Chem Lab Med . 2016 апр. 54 (4): 627-32. [Медлайн].
Хиршхорн Р., Канотти Ф. Иммунодефицит из-за нарушений пуринового обмена. Ochs HD, Smith CIE, Puck JM, ред. Первичные иммунодефицитные заболевания: молекулярно-генетический подход . 2-е изд. Нью-Йорк, штат Нью-Йорк: Oxford University Press, Inc; 2007. 169-96.
Хершфилд М.С., Митчелл Б.С. Иммунодефицитные заболевания, вызванные дефицитом аденозиндезаминазы и пуриновой нуклеозидфосфорилазы.Скривер Ч.Р., Боде А.Л., Слай В.С., Валле Д. Метаболические и молекулярные основы наследственного заболевания . Нью-Йорк: Макгроу-Хилл; 2001. 2585-2625.
Arpaia E, Benveniste P, Di Cristofano A, et al. Митохондриальная основа иммунодефицита. Данные, полученные на мышах с дефицитом пуриновой нуклеозид-фосфорилазы. J Exp Med . 2000, 19 июня. 191 (12): 2197-208. [Медлайн].
Bzowska A, Kulikowska E, Shugar D. Пуриновые нуклеозидфосфорилазы: свойства, функции и клинические аспекты. Pharmacol Ther . 2000 декабрь 88 (3): 349-425. [Медлайн].
Markert ML, Hershfield MS, Schiff RI, Buckley RH. Дефицит аденозиндезаминазы и пуриннуклеозидфосфорилазы: оценка терапевтических вмешательств у восьми пациентов. Дж. Клин Иммунол . 1987 Сентябрь 7 (5): 389-99. [Медлайн].
Маркерт МЛ. Дефицит пуриновой нуклеозидфосфорилазы. Иммунодефицитная Ред. . 1991. 3 (1): 45-81. [Медлайн].
Хиршхорн Р.Обзор биохимических аномалий и молекулярной генетики дефицита аденозиндезаминазы. Педиатр Рес . 1993. 33: S35-41.
Hershfield MS. Дефицит аденозиндезаминазы: клинические проявления, молекулярные основы и терапия. Семин Гематол . 1998. 35: 291-298.
Дефицит пуриновой нуклеозидфосфорилазы. Домашний справочник по генетике. Доступно по адресу https://ghr.nlm.nih.gov/condition/purine-nucleoside-phosphorylase-deficiency.2019 июл 16; Доступ: апрель 2019 г.
Дефицит пуриновой нуклеозидфосфорилазы. Сиротка. Доступно на https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=760. Доступ: апрель 2015 г.
Aytekin C, Dogu F, Tanir G, et al. Дефицит пуриновой нуклеозидфосфорилазы со смертельным исходом у двух сестер. Eur J Педиатр . 2010 Март 169 (3): 311-4. [Медлайн].
Parvaneh N, Ashrafi MR, Yeganeh M, Pouladi N, Sayarifar F, Parvaneh L.Прогрессирующая мультифокальная лейкоэнцефалопатия при дефиците пуриновой нуклеозидфосфорилазы. Brain Dev . 2007 марта 29 (2): 124-6. [Медлайн].
Брэдфорд К.Л., Моретти Ф.А., Карбонаро-Саррачино Д.А., Гаспар Н.Б., Кон Д.Б. Тяжелый комбинированный иммунодефицит с дефицитом аденозиндезаминазы (ADA) (SCID): молекулярный патогенез и клинические проявления. Дж. Клин Иммунол . 2017 окт.37 (7): 626-637. [Медлайн].
Мэнсон Д., Даймонд Л., Уджхан К., Хуссейн Ф. Б., Ройфман С., Грюнебаум Э.Характерные изменения лопатки и ребер на рентгенограммах грудной клетки детей с ADA-дефицитом SCIDS в первый год жизни. Педиатр-Радиол . 2013 Март 43 (5): 589-92. [Медлайн].
Грюнебаум Э, Катц Э, Ройфман СМ. Легочный альвеолярный протеиноз у пациентов с дефицитом аденозиндезаминазы. J Allergy Clin Immunol . 2012 июн 129 (6): 1588-93. [Медлайн].
Hirschhorn R. Возвращение к нормальному состоянию наследственных мутаций у людей in vivo. Дж. Мед Генет . 2003. 40: 721-728.
Routes JM, Grossman WJ, Verbsky J, et al. Скрининг новорожденных на тяжелую Т-клеточную лимфопению в масштабе штата. ЯМА . 2009 декабрь 9. 302 (22): 2465-70. [Медлайн].
Грюнебаум Э, Чжан Дж., Ройфман СМ. Новые мутации и горячие точки у пациентов с дефицитом пуриновой нуклеозидфосфорилазы. Нуклеозиды Нуклеотиды Нуклеиновые кислоты . 2004 23 октября (8-9): 1411-5. [Медлайн].
[Рекомендации] Bonilla FA, Bernstein IL, Khan DA, et al.Параметр практики для диагностики и лечения первичного иммунодефицита. Ann Allergy Asthma Immunol . 2005 г., май. 94 (5 доп. 1): S1-63. [Медлайн].
Кон ДБ, Гаспар НВ. Как мы справляемся с тяжелым комбинированным иммунодефицитом с дефицитом аденозиндезаминазы (ADA SCID). Дж. Клин Иммунол . 2017 май. 37 (4): 351-356. [Медлайн].
Hassan A, Booth C, Brightwell A, et al. Результат трансплантации гемопоэтических стволовых клеток при тяжелом комбинированном иммунодефиците с дефицитом аденозиндезаминазы. Кровь . 2012 окт 25.120 (17): 3615-24; викторина 3626. [Medline].
Флинн AM, Геннери АР. Дефицит аденозиндезаминазы: обзор. Орфанет Дж. Редкий Диск . 2018 24 апреля. 13 (1): 65. [Медлайн]. [Полный текст].
Broome CB, Graham ML, Saulsbury FT и др. Коррекция дефицита пуриновой нуклеозидфосфорилазы путем трансплантации аллогенного костного мозга от брата или сестры. Дж. Педиатр . 1996 марта 128 (3): 373-6.[Медлайн].
Майерс Л.А., Хершфилд М.С., Нил В.Т. и др. Дефицит пуриновой нуклеозидфосфорилазы (PNP-def), проявляющийся лимфопенией и задержкой развития: успешная коррекция с помощью трансплантации пуповинной крови. Дж. Педиатр . 2004 ноябрь 145 (5): 710-2. [Медлайн].
Classen CF, Schulz AS, Sigl-Kraetzig M, et al. Успешная трансплантация HLA-идентичного костного мозга пациенту с дефицитом PNP с использованием бусульфана и флударабина для кондиционирования. Пересадка костного мозга . 2001 июля 28 (1): 93-6. [Медлайн].
О’Рейли Р.Дж., Кивер С., Кернан Н.А. и др. Трансплантация костного мозга с неидентичными T-клетками HLA: сравнение результатов у пациентов, получавших лечение от лейкемии и тяжелого комбинированного иммунодефицита. Протокол трансплантологии . 1987, 19 декабря (6, приложение 7): 55-60. [Медлайн].
Фишер А., Гриселли К. [Трансплантат костного мозга: реакция трансплантата против хозяина и отторжение]. Нефрология .1986. 7 (3 Suppl): 1-4. [Медлайн].
Hershfield MS. Ферментативная заместительная терапия дефицита аденозиндезаминазы с модифицированной полиэтиленгликолем аденозиндезаминазой (PEG-ADA). Иммунодефицит . 1993. 4 (1-4): 93-7. [Медлайн].
Toro A, Paiva M, Ackerley C, Grunebaum E. Внутриклеточная доставка пуриновой нуклеозидфосфорилазы (PNP), слитой с доменом трансдукции белка, корректирует дефицит PNP in vitro. Клеточный иммунол .2006 апр. 240 (2): 107-15. [Медлайн].
Кон Д.Б., Хершфилд М.С., Карбонаро Д. и др. Т-лимфоциты с нормальным геном ADA накапливаются после трансплантации трансдуцированных аутологичных CD34 + клеток пуповинной крови новорожденным с ТКИД с дефицитом ADA. Нат Мед . 1998 июл.4 (7): 775-80. [Медлайн].
Айути А., Славин С., Акер М. и др. Коррекция ADA-SCID с помощью генной терапии стволовыми клетками в сочетании с немиелоаблативным кондиционированием. Наука .2002, 28 июня, 296 (5577): 2410-3. [Медлайн].
Айути А., Каттанео Ф, Галимберти С. и др. Генная терапия иммунодефицита вследствие дефицита аденозиндезаминазы. N Engl J Med . 2009 29 января. 360 (5): 447-58. [Медлайн].
Cicalese MP, Ferrua F, Castagnaro L, Rolfe K, De Boever E, Reinhardt RR, et al. Генная терапия дефицита аденозиндезаминазы: всесторонняя оценка краткосрочной и среднесрочной безопасности. Мол тер .2018 7 марта. 26 (3): 917-931. [Медлайн]. [Полный текст].
Купер А.Р., Лилль Г.Р., Шоу К., Карбонаро-Саррачино Д.А., Давила А., Соколич Р. и др. Интенсивность циторедуктивного кондиционирования предсказывает клональное разнообразие у пациентов с ретровирусной генной терапией ADA-SCID. Кровь . 2017 г. 11 мая. 129 (19): 2624-2635. [Медлайн]. [Полный текст].
Ляо П., Торо А., Мин В., Ли С., Ройфман С.М., Грунебаум Э. Генная терапия лентивирусом при дефиците пуриновой нуклеозидфосфорилазы. Дж. Джин Мед . 2008 г. 10 (12): 1282-93. [Медлайн].
Bonagura VR, Marchlewski R, Cox A, Rosenthal DW. Биологический уровень IgG при первичном иммунодефицитном заболевании: уровень IgG, который защищает от рецидивирующей инфекции. J Allergy Clin Immunol . 2008 июл.122 (1): 210-2. [Медлайн].
Durandy A, Wahn V, Petteway S, Gelfand EW. Заместительная иммуноглобулиновая терапия при заболеваниях с первичной недостаточностью антител — максимальный успех. Int Arch Allergy Immunol . 2005. 136: 217-229.
Гарсия-Льорет М., МакГи С., Шатила Т.А. Заместительная иммуноглобулиновая терапия у детей. Immunol Allergy Clin North Am . 2008, 28 ноября (4): 833-49, ix. [Медлайн]. [Полный текст].
Hooper JA. Внутривенные иммуноглобулины: эволюция коммерческих препаратов ВВИГ. Immunol Allergy Clin North Am . 2008, 28 ноября (4): 765-78, viii. [Медлайн].
Шах С.Рекомендации по применению IGIV в аптеке. Am J Health Syst Pharm . 2005 15 августа 62 (16 Дополнение 3): S5-11. [Медлайн].
Markert ML, Finkel BD, McLaughlin TM, et al. Мутации при дефиците пуриновой нуклеозидфосфорилазы. Хум Мутат . 1997. 9: 118-121.
Бакли Р. Заболевания первичного иммунодефицита из-за дефекта лимфоцитов. N Engl J Med . 2 ноября 2000 г. 343 (18): 1313-24. [Медлайн].
Бакли Р. Х., Шифф Р. И., Шифф С. Е. и др.Тяжелый комбинированный иммунодефицит человека: генетическое, фенотипическое и функциональное разнообразие у ста восьми младенцев. Дж. Педиатр . 1997 Mar.130 (3): 378-87. [Медлайн].
Джоши А.Ю., Айер В.Н., Хаган Дж.Б., Сент-Совер Дж.Л., Бойс Т.Г. Заболеваемость и временные тенденции первичного иммунодефицита: популяционное когортное исследование. Mayo Clin Proc . 2009. 84 (1): 16-22. [Медлайн]. [Полный текст].
Lacy CF, Armstrong LL, Goldman MP, Lance LL, ред. Справочник по лекарствам . Кливленд, Огайо: Lexi-Comp, Inc; 2009.
Siegel J. Продукт: Все иммуноглобулины для внутривенного введения не эквивалентны. Фармакотерапия . 2005 25 ноября (11, часть 2): 78С-84С. [Медлайн].
границ | Частичный дефицит пуриновой нуклеозидфосфорилазы помогает определить минимальную активность, необходимую для иммунного и неврологического развития
Введение
Пуриннуклеозидфосфорилаза (PNP) представляет собой фермент 32 килодальтон (кДа), который обратимо катализирует фосфолиз инозина, дезокси-инозина, гуанозина и дезоксигуанозина (рис. 1) (1).Полное или почти полное отсутствие PNP вызывает накопление субстратов фермента в крови и моче, предотвращая при этом образование продуктов фермента, гипоксантина, ксантина и, в конечном итоге, мочевой кислоты (2, 3). Кроме того, увеличивается концентрация фосфорилированных производных гуанозина и дезоксигуанозина, GTP и dGTP соответственно. Накопление dGTP в митохондриях клеток препятствует поддержанию и репарации ДНК (2). Последнее приводит к усилению апоптоза клеток, подвергающихся быстрой репликации, или клеток, подвергшихся окислительному и лучевому стрессу (2, 4), аналогично снижению выживаемости клеток с мутированным геном ataxia telangiectasia (5).
Рисунок 1 . Роль PNP в метаболизме пуринов и последствия дефицита PNP. PNP обратимо катализирует фосфолиз инозина, дезокси-инозина, гуанозина и дезоксигуанозина. Дефицит PNP вызывает накопление (показано направленными вверх стрелками) субстратов ферментов и их фосфорилированных производных, GTP и dGTP, предотвращая при этом образование (показано стрелками, направленными вниз) гипоксантина, гуанина, ксантина и, впоследствии, мочевой кислоты. .Ферменты отмечены серым фоном.
Аутосомно-рецессивные дефекты ферментов PNP были обнаружены в 1970-х годах среди пациентов с глубоким Т-клеточным иммунодефицитом (6). Подобно другому наследственному пуриновому дефекту, дефицит аденозиндезаминазы (ADA), прогрессирующие аномалии в субпопуляциях лимфоцитов B и Natural Killer (NK), а также в миелоидной линии также выявляются у пациентов с дефицитом PNP (4, 7). Пациенты с дефицитом PNP часто страдают бактериальными, вирусными, грибковыми или оппортунистическими инфекциями в первый год жизни (8, 9).Иммунная дисрегуляция, связанная с дефицитом PNP, также приводит к аутоиммунитету, что приводит к гематологической цитопении и частым злокачественным трансформациям (10, 11). Неинфекционная дисфункция, особенно неврологические аномалии, такие как атаксия и спастичность, часто наблюдаются у пациентов с дефицитом PNP (12) и у мышей, лишенных фермента PNP (13). Эти аномалии могут быть связаны с повсеместной природой фермента PNP и разнообразной биологической ролью PNP и пуриновых метаболитов.
Антимикробное лечение и профилактика могут предотвратить некоторые инфекции, вызванные дефицитом PNP, однако большинство пациентов умирают от инфекций, аутоиммунных заболеваний или злокачественных новообразований в младенчестве и раннем детстве (14–16).Трансплантация аллогенных гемопоэтических стволовых клеток (HSC) может исправить иммунный дефицит и, возможно, остановить или даже улучшить неврологический дефицит. Тем не менее, осложнения, связанные с трансплантатом, все еще часты, и типичное (нормальное) развитие не всегда достигается, возможно, из-за неполного донорского химеризма, который ограничивает способность PNP-опытных гемопоэтических клеток восстанавливать пуриновый метаболизм (8, 12, 17–21). Более того, даже при достижении донорского химеризма неврологический дефицит может сохраняться.Эти аномалии могут быть связаны с необратимым пре-трансплантационным повреждением или необходимостью активности PNP в нейрональных клетках, что также наблюдается у некоторых пациентов с дефицитом ADA после трансплантации HSC (22, 23).
Альтернативные методы лечения, которые изучаются при дефиците PNP, включают инъекции замены фермента PNP человеческим PNP, слитым с доменом трансдукции белка (TATPNP) (24), или генную терапию с использованием гена PNP, доставленного ex-vivo в аутологичный PNP-дефицитный HSC (25).Хотя эти методы лечения в настоящее время недоступны для клинического использования, определение минимальной активности PNP, необходимой для коррекции иммунных и неврологических нарушений, связанных с дефицитом PNP, будет важным шагом для разработки и оценки потенциальных будущих клинических методов лечения.
Мы предположили, что пациенты с частичным дефицитом PNP, характеризующиеся поздним и легким фенотипом из-за остаточных ферментов PNP, могут предоставить ценную информацию об уровнях PNP, которых будет достаточно для нормального или близкого к нормальному иммунного и неврологического развития.
Здесь мы описываем трех братьев и сестер с дефицитом PNP, которые были идентифицированы в зрелом возрасте с легкими-умеренными иммунными аномалиями и типичной неврологической функцией.
Материалы и методы
Пациенты
Были изучены трое братьев и сестер с гомозиготной мутацией c.769C> G в гене PNP . Все исследования проводились в соответствии с советами по этике исследований Госпитального центра Монреальского университета, Монреаль, Квебек, и Больницы для больных детей, Торонто, Онтарио.
Оценка иммунитета
Проточная цитометрияиспользовалась для подсчета субпопуляций лимфоцитов и процентного содержания 24 семейств V-бета среди Т-клеток CD4 + и CD8 + с помощью анализа репертуара Т-клеток IOTest ® Beta Mark (Beckman Coulter, IND, США. ), как описано ранее (26). Пролиферацию мононуклеарных клеток периферической крови (PBMC) определяли после 4-дневной стимуляции фитогемагглютинином (PHA) включением 3 [H] тимидина. Индекс стимуляции (SI) рассчитывали как отношение включения 3 [H] тимидина в клетки со стимуляцией или без нее.Верхний предел SI для пациентов с комбинированным иммунодефицитом составляет 241,3. Пролиферацию жизнеспособных синглетных клеток CD4 + T после пятидневной стимуляции антиCD3 (5 мкг / мл) и растворимым антиCD28 (2 мкг / мл) также измеряли проточной цитометрией с использованием внутриклеточного разведения фиолетового красителя (CellTrace ™ Violet от Thermofisher ).
Активность фермента PNP
АктивностьPNP, выраженная в процентах от здорового контроля того же дня, была измерена полуколичественно, как описано ранее, путем преобразования [8-14C] инозина в гипоксантин, разделенного тонкослойной хроматографией (27) с небольшими изменениями, как результаты не были нормализованы по содержанию белка или гемоглобина в образцах.Активность PNP измеряли в гемолизатах, содержащих преимущественно эритроциты, в PBMC и в лимфобластоидных B-клетках, трансформированных EBV, установленных, как описано ранее (28). Активность PNP in vivo оценивалась путем измерения мочевой кислоты в крови, гуанозина и дезоксигуанозина в сухих пятнах крови, как недавно описано (29), и субстратов PNP в моче с использованием жидкостной хроматографии и тандемной масс-спектрометрии.
Экспрессия белка PNP
ЭкспрессиюPNP определяли вестерн-блоттингом, как описано ранее (30), в лизатах 5 × 10 6 лимфобластоидных клеток пациента II.1, пациент с отсутствующей активностью PNP (c.172C> T, pArg57Ter и 285 + 1 G> A), о котором сообщалось ранее (31), пациент с дефицитом ADA (c. 646 G> A, p. Gly216Arg и c. 955delGAGAA) и здоровый контроль. Кроме того, блотировали 100 нг TATPNP с молекулярной массой ~ 34 кДа (30) и маркеры молекулярной массы. После разделения белков и переноса на нитроцеллюлозную мембрану (Biorad) добавляли разбавленное 1: 1000 моноклональное мышиное антитело против человеческого PNP (LS-C137543 от Life-Span Bio Inc, Сиэтл, Калифорния).Белки детектировали с помощью соответствующих вторичных антител и реагентов для определения Вестерн-блоттинга Amersham ECL (GE Healthcare, Чикаго, Иллинойс). Мембраны были повторно заполнены бета-актином. Интенсивность полос PNP и бета-актина определяли с помощью программного обеспечения открытой платформы IMAGEJ и рассчитывали соотношение между полосами.
Ремонт ДНК
ВосстановлениеДНК определяли путем измерения выживаемости лимфобластоидных клеток (0,5 × 10 6 / мл ) через 3 дня после ионизирующего облучения 10 Гр, как описано ранее (32).Выживаемость клеток, установленная у пациентов II.1 и II.2, пациента с отсутствующей активностью ПНФ, описанной выше, двух несвязанных с ПНП контрольных групп и пациента с мутированным геном ataxia telangiectasia (c. 4642delGATA и c. 5932G > T, p Glu1978Ter) сравнивали. Фракцию выживания (SF) рассчитывали путем деления количества жизнеспособных клеток после облучения на начальное количество жизнеспособных клеток в необлученных культурах. Статистические различия в SF определялись с помощью двухфакторного анализа Anova и считались значимыми, если p <0.05.
Результаты
Пациенты
Пациентка II.1 (семейная родословная изображена на рис. 2А), 21-летняя студентка университета, была направлена на обследование на рецидивирующие сино-легочные инфекции и стойкую лимфопению. Пациентка перенесла пневмонию в 8 и 20 лет, ветряную оспу в 4 года (вакцину против ветряной оспы не получала) и опоясывающий лишай в 19 лет. Она также страдала пансинуситом и перенесла полипэктомию носа в 20 лет.У пациента был эпизод преходящей нейтропении, связанной с пневмонией, который разрешился после короткого курса введения гранулоцитарного колониестимулирующего фактора. Она прошла вакцинацию против кори, эпидемического паротита и краснухи без каких-либо осложнений и не болела оральным молочницей или другими грибковыми инфекциями. Она была младшей из шести детей кровных родителей. Физикальное обследование пациента, включая подробное неврологическое обследование, было нормальным. У пациентки развилась астма без признаков бронхоэктазии при рентгенологическом исследовании грудной клетки.После постановки диагноза иммунодефицит, а затем гипогаммаглобулинемии, пациентка была пролечена профилактическим введением триметоприм-сульфаметоксазола и иммуноглобулина с временным улучшением заболевания носовых пазух. ДНК, выделенная из PBMC пациента и протестированная с помощью коммерческой панели (Blueprint Genetics ® ) на 232 гена, связанных с первичным иммунодефицитом (PID), выявила гомозиготный вариант c.769C> G в экзоне 6 гена PNP . Впоследствии присутствие мутации было подтверждено секвенированием по Сэнгеру (рис. 2В, верхняя панель).Ранее сообщалось об этом варианте у пациента с дефицитом ПНФ с комплексными гетерозиготными мутациями (33). Вариант предположительно заменил умеренно консервативный гистидин в положении 257 на аспарагиновую кислоту (p.His257Asp). In silico Анализ с помощью PolyPhen, SIFT и MutationTaster предсказал, что этот вариант является вредоносным и вызывает заболевания. Мутации, вызывающие заболевание, в других потенциальных генах PID не идентифицированы. Гомозиготная мутация была также идентифицирована у братьев и сестер II.2 (рис. 2В, средняя панель) и подтверждена секвенированием по Сэнгеру.Родители и брат или сестра пациентов II.4 были гетерозиготными по данному варианту.
Рисунок 2 . Семейная родословная и секвенирование гена PNP пациентов II.1 и II.2. (A) Семейная родословная, демонстрирующая трех братьев и сестер (II.1, II.2 и II.3), которые являются гомозиготными по мутации гена PNP , предположительно вызывающей p.His257Asp (обозначенной как h357D), в то время как родители ( I.1 и I.2) и дополнительный сиблинг (II.4) были гетерозиготными по мутированным аллелям и аллелям дикого типа (обозначенным как WT). (B) Секвенирование гена PNP Секвенирование по Сэнгеру гена PNP от пациентов II.1 (верхняя панель) и II.2 (средняя панель), демонстрирующее нуклеотид G в положении 769, в то время как здоровый контроль ( нижняя панель) имеет нуклеотид C. Секвенирование выполняли с прямым праймером TTACAGGTGTGAACCACTGC и обратным праймером GAAGAAAGTGGGAAAGGTGA.
Пациент II.2, 25-летний мужчина, переболел ветряной оспой без осложнений в возрасте 2 лет (он не получал вакцину от ветряной оспы) и не сообщил о каких-либо серьезных инфекциях, кроме инфекции раны колена, после аварии на скутере в 16 лет. лет и бородавки в 25 лет.Он работал оператором тяжелой техники. Пациент II.3, 28-летний мужчина, имел несколько инфекций верхних дыхательных путей в раннем детстве, тяжелую ветряную оспу в 5-летнем возрасте, опоясывающий лишай в 6,5-летнем возрасте и пневмонию в 10-летнем возрасте. Сообщалось, что в подростковом возрасте у него были проблемы с мелкой моторикой и координацией. Оба пациента II.2 и II.3 не страдали аутоиммунными заболеваниями или злокачественными новообразованиями, и их физикальное обследование, включая подробное неврологическое обследование, было нормальным.Родители пациентов и трое оставшихся братьев и сестер не страдали рецидивирующими инфекциями, аутоиммунными заболеваниями или злокачественными новообразованиями, хотя отец (I.1) был исследован на предмет воспалительного узелка в легких.
Оценка иммунитета
Количество лимфоцитов было почти нормальным у пациента II.2 и умеренно сниженным у пациентов II.1 и II.3, с нормальными числами лейкоцитов и нейтрофилов у трех братьев и сестер (Таблица 1). У пациентов II.1 и II.3 также было умеренно сниженное количество CD4 + и CD8 + Т-клеток, CD19 + В-клеток и CD3 — CD16 + CD56 + NK-клеток в сравнении. здоровому контролю того же возраста (34), в то время как количество субпопуляций лимфоцитов пациента II.2 были нормальными или почти нормальными. У обоих пациентов II.1 и II.2 было пониженное количество наивных Т-клеток, в то время как круги удаления рецепторов Т-клеток не были обнаружены. Процентное содержание CD3 + CD4 + CD25 + CD127 Low регуляторных Т-клеток было нормальным. Разнообразие Т-клеток пациента II.1 было ограничено, особенно для Т-клеток CD8 + , где 8 из 24 семейств V-бета отсутствовали (рис. 3А). Пациенты II.1 и II.2 PBMC продемонстрировали нормальный SI до PHA. Аналогично пациенты II.1 и II.2 CD4 + Т-клетки продемонстрировали нормальную пролиферацию, как определено анализом проточной цитометрии разведения фиолетового красителя (рис. 3В). Пациентка II.1 изначально имела повышенный уровень IgG до 28,5 г / л, который снизился до 4,2 г / л после эффективного лечения синусита. У пациента II.3 также была гипогаммаглобулинемия, тогда как у пациента II.2 был нормальный IgG. У пациента II.1 не было антител к столбняку или дифтерии, несмотря на предыдущие вакцинации, хотя у пациентов II.1 и II.2 были антитела к EBV.Пациент II.3 имел антитела к гепатиту B после вакцинации. У пациента II.1 также был повышен уровень антиядерных антител до 1: 640, тогда как у пациентов II.1 и II.3 антитела к двухцепочечной ДНК и экстрагируемые ядерные антигены не были обнаружены, а уровни комплемента C3 и C4 были нормальными.
Таблица 1 . Лабораторные исследования пациентов с частичной недостаточностью PNP.
Рисунок 3 . Оценка иммунитета у пациентов с частичной недостаточностью PNP. (A) CD8 + Разнообразие Т-клеток у пациента II.1. Разнообразие Т-клеток определяли с помощью проточной цитометрии как процент CD3 + CD8 + Т-клеток, экспрессирующих одну из 24 V-бета-цепей TCR. Разнообразие пациентов II.1 изображено черными столбцами, а среднее значение и 1 стандартное отклонение для здоровых доноров представлены белыми столбцами. (B) Пролиферация CD4 + Т-клеток пациентов с частичной недостаточностью PNP. Ответ CD4 + Т-клеток от пациента II.1 (светло-фиолетовый) и II.2 (светло-розовый) после стимуляции анти-CD3 и растворимым анти-CD28 в течение 5 дней измеряли с помощью разведения внутриклеточного фиолетового красителя с помощью проточной цитометрии и сравнивали с контрольным здоровьем в тот же день (желтый).
Оценка PNP
ДефицитPNP обычно вызывает накопление в крови и моче субстратов фермента инозина, гуанозина, дезокси-инозина и дезоксигуанозина, а также к снижению или отсутствию образования мочевой кислоты. Пациенты II.1 и II.2 имели нормальную концентрацию гуанозина и дезоксигуанозина в крови.Точно так же мочевая кислота в крови II.1, II.2 и II.3 была в пределах нормы. Напротив, субстраты PNP, не обнаруженные в моче здоровых людей, имеющих опытный PNP, были повышены в моче пациентов II.1, II.2 и II.3 (Таблица 2).
Таблица 2 . Активность PNP у пациентов с частичным дефицитом PNP.
Активность PNP в гемолизатах обычно отсутствует или заметно снижена у пациентов с дефицитом PNP. Напротив, у пациентов II он составлял 182 и 166 нмоль / мин / мл.1 и II.2, соответственно (10,7 и 8,9%, соответственно, здорового контроля в тот же день) (таблица 2). Активность ADA в гемолизатах пациентов II.1 и II.2, измеренная для обеспечения целостности образца крови, была нормальной и составляла 104 и 75 нмоль / мин / мл соответственно. Подобно результатам гемолизатов, активность PNP в PBMC пациентов II.1 и II.2 была снижена на 0,13 и 014 нмоль / мин / 10 6 клеток, соответственно (8,8 и 9,1% от здорового контроля в тот же день, соответственно). Из-за потенциальной вариабельности измерений образцов отдельных пациентов мы также измерили активность PNP в клеточных линиях пациентов, что позволило проводить повторные оценки и сравнение с несколькими контрольными образцами одновременно (таблица 2).Активность ПНФ у пациентов II.1 и II.2 составила 8,4 ± 2,8% и 10,5 ± 1,3% от здоровых контролей (3 независимых эксперимента).
Вестерн-блоттинг проводили на лизатах лимфобластоидных клеток, трансформированных EBV, для оценки влияния мутации гена PNP на экспрессию его белка (фиг. 4A). В то время как белок PNP не был обнаружен в лимфобластоидных клетках, полученных от пациента с отсутствующим PNP, ожидаемая полоса 32 кДа была видна на дорожках, соответствующих лизатам, полученным от здорового контроля, владеющего PNP, и пациента с дефицитом ADA.Аналогичным образом, полоса 32 кДа была обнаружена в лимфобластоидных клетках, полученных от пациента II.1. Также ожидаемая полоса 34 кДа наблюдалась для дорожки, загруженной TATPNP. Полуколичественная оценка, путем сравнения отношения PNP к бета-актину и интенсивности полосы для TATPNP, показала, что количество PNP в клетках пациента II.1 составляло ~ 50 нг и аналогично количеству PNP-опытных клеток (рис. 4B). ).
Рисунок 4 . Экспрессия белка PNP в клетках пациента с частичным дефицитом PNP. (A) Вестерн-блоттинг лимфобластоидных клеток, трансформированных EBV, от пациента II.1, пациента с полным дефицитом PNP, здорового контроля со знанием PNP и пациента с дефицитом ADA. Также блотировали TATPNP и PNP с молекулярной массой 34 и 32 кДа соответственно, и маркеры молекулярной массы. (B) Полуколичественная оценка белка PNP в лимфобластоидных клетках, трансформированных EBV, от пациентов и контрольной группы, описанных выше, была рассчитана путем сравнения с интенсивностью полосы бета-актина и TATPNP с использованием программного обеспечения открытой платформы IMAGEJ.
Чувствительность лимфобластоидных клеток к облучению
Пуриновые метаболиты важны для репликации и репарации ДНК, а аномальный пуриновый гомеостаз может повышать чувствительность клеток к ионизирующему облучению (4). Соответственно, жизнеспособность лимфобластоидных клеток определялась через 3 дня после облучения дозой 10 Гр по сравнению с начальным количеством клеток до облучения (фиг. 5). Как и ожидалось, лимфобластоидные клетки, полученные от пациента с мутированным геном ataxia telangiectasia и пациента с полным отсутствием активности PNP, были особенно чувствительны к облучению, что привело к значительному снижению (p <0.001) SF по сравнению с контролями. Напротив, клетки, полученные от пациентов II.1 и II.2, имели SF, который статистически не отличался от PNP-опытного контроля, но значительно увеличился по сравнению с клетками от пациентов с атаксией, телеангиэктазией и отсутствием активности PNP.
Рисунок 5 . Чувствительность клеток пациентов с частичным дефицитом PNP к облучению. Жизнеспособность лимфобластоидных клеток пациентов II.1 и II.2, пациента с полным отсутствием активности ПНФ, пациента с атаксией-телеангиэктазией и двух несвязанных контрольных групп, не обладающих эффективностью ПНФ, определяли через 3 дня после облучения в дозе 10 Гр.Фракцию выживания (SF) рассчитывали путем деления количества жизнеспособных клеток после облучения на количество жизнеспособных клеток в необлученных культурах. Статистические различия в SF определяли с помощью двухкомпонентного Anova.
Обсуждение
Здесь мы сообщаем об идентификации и характеристике трех братьев и сестер на третьем десятилетии жизни с гомозиготной мутацией гена His257Asp PNP , приводящей к частичному дефициту PNP. Ранее о дефиците ПНФ сообщалось менее чем у 80 пациентов, у многих из них был тяжелый иммунодефицит в младенчестве, и лишь некоторые дожили до взрослого возраста (35).Среди более длительно выживших пациентов с ПНП — 21-летняя беременная женщина с отсутствием ПНП, которая оставалась практически здоровой с момента ее обращения в 3-летнем возрасте, но лечилась профилактическими антибиотиками и иммуноглобулинами (9). Она также страдала атаксией, спастичностью и значительной задержкой умственного развития. Другая пациентка с дефицитом PNP была здорова до 6 лет, когда у нее развилась тяжелая аутоиммунная цитопения, за которой последовал синусит и приступ ветрянки со смертельным исходом в 11 лет (35).Третий пациент с «поздним началом» дефицита PNP с очень низкой активностью PNP страдал от рецидивирующих респираторных инфекций, приводящих к бронхоэктазии, и его лечили внутривенными иммуноглобулинами до трансплантации HSC в возрасте 13 лет (36). Напротив, среди трех братьев и сестер, описанных здесь, у одного пациента не было инфекций и был почти нормальный иммунитет в возрасте 25 лет, в то время как у двух других пациентов были только умеренные инфекции и лабораторные отклонения, которые не потребовали значительного медицинского вмешательства в первые два года. десятилетия жизни.Кроме того, ни один из братьев и сестер не страдал аутоиммунитетом или злокачественными новообразованиями, которые распространены среди пациентов с дефицитом PNP (35). Таким образом, насколько нам известно, описанные здесь братья и сестры с частичным дефицитом PNP являются самыми старыми людьми, у которых выявлена аномальная активность PNP и самый мягкий фенотип, что еще больше увеличивает возраст проявления и клинические проявления, связанные с этим редким дефектом.
Описанные здесь пациенты демонстрируют дополнительный аспект диагностики недостаточности PNP.Гипоурикемия считается возможным ключом к диагностике дефицита PNP у пациентов с ВЗОМТ (37). Тем не менее, аналогично недавнему отчету, в котором была обнаружена нормальная мочевая кислота у пяти из семи лиц с дефицитом PNP (33), описанные здесь пациенты имели нормальную мочевую кислоту в крови, а у двух из них также были нормальные субстраты PNP в крови, возможно, из-за остаточная активность PNP. Следовательно, вместо мочевой кислоты и метаболитов пурина в крови измерение субстратов PNP в моче может быть более чувствительным методом выявления аномальной функции PNP, хотя во многих центрах такой тест недоступен.
Среди братьев и сестер, несущих одну и ту же мутацию гена PNP, наблюдались некоторые расхождения в степени тяжести инфекций и иммунных аномалий. Один из братьев и сестер страдал рецидивирующими сино-легочными инфекциями и нуждался в заместительной терапии иммуноглобулином, в то время как у двух других братьев и сестер не было симптомов в течение многих лет. Хотя активность PNP измерялась только в клетках пациентов II.1 и II.2, сходная нормальная мочевая кислота в крови и повышенные уровни субстратов PNP в моче, наблюдаемые у трех братьев и сестер, позволяют предположить, что все они обладают одинаковой активностью фермента PNP.Следовательно, вполне вероятно, что фенотипические различия между братьями и сестрами связаны не с измеренным уровнем активности фермента, а скорее с еще нераспознанными внешними или внутренними факторами, как уже было продемонстрировано для других моногенных первичных иммунодефицитов (38).
Для дальнейшего изучения биологического значения остаточной активности PNP, обнаруженной у пациентов, мы протестировали ex vivo функций, которые зависят от PNP, включая пролиферацию клеток и чувствительность к облучению (2, 4, 39).Примечательно, что пролиферация Т-клеток пациентов II.1 и II.2 при стимуляции была нормальной, что также может объяснить их способность выживать при ветряной оспе, о которой обычно сообщают как о разрушительной инфекции среди пациентов с дефицитом PNP (8, 17, 35, 36, 40). Точно так же выживаемость лимфобластоидных В-клеток пациентов II.1 и II.2 при ионизирующем облучении была нормальной и значительно увеличилась по сравнению с пациентом без PNP (4). Важно отметить, что способность поддерживать защитный иммунитет только при 8–11% активности PNP предполагает, что это может быть достаточной пороговой активностью, необходимой для лечения дефицита PNP с помощью текущих аллогенных HSC или будущих аутологичных трансплантатов HSC и заместительной ферментативной терапии.
Неврологический дефицит, особенно тонус и двигательные нарушения, встречаются у многих пациентов с дефицитом PNP (1). Напротив, у трех братьев и сестер, описанных здесь, было типичное (нормальное) неврологическое развитие, возможно, из-за остаточной активности PNP в их нейрональных клетках. Предполагается, что причиной этих нарушений является ускоренный апоптоз нейрональных клеток с дефицитом PNP (13). Дополнительным доказательством этой гипотезы является обнаруженная здесь корреляция между неврологическим статусом и устойчивостью клеток к облучению.Клетки пациента с мутированным геном ataxia telangiectasia и пациента с отсутствующей активностью PNP, который страдал атаксией и замедленными двигательными навыками, продемонстрировали заметно снижение выживаемости после облучения. Напротив, клетки неврологически интактных пациентов, описанных здесь, имели нормальную выживаемость после облучения. Это открытие дополнительно подтверждает наше предыдущее наблюдение, что достижение 10–20% нормальной активности PNP в клетках мозга может предотвратить неврологические нарушения, влияющие на PNP-дефицитных мышей (13).
Информация, полученная от пациентов с дефицитом PNP и уникальными мутациями, использовалась для разработки аналоговых ингибиторов PNP в переходном состоянии (41). Такие ингибиторы PNP были исследованы как потенциальные средства лечения неконтролируемой пролиферации Т- и В-клеток, наблюдаемой при лейкемии, аутоиммунных заболеваниях, отторжении трансплантата и болезни трансплантат против хозяина (41, 42). Гистидин в положении 257 молекулы PNP напрямую взаимодействует с субстратами фермента, однако существуют противоречивые данные относительно его роли в каталитическом механизме PNP (43, 44), отсюда и интерес к биохимическим эффектам мутантного His257 in vivo .Ранее сообщалось о мутации His257Asp у 19-летнего пациента с дефицитом PNP вместе с вариантом 172C> T, который предположительно вызвал преждевременную остановку (33). Мутации соединения PNP привели к нормальной мочевой кислоте крови, несмотря на отсутствие активности фермента, однако дополнительные клинические и иммунологические данные не были предоставлены, что ограничивает возможность оценить влияние мутации His257. Поскольку описанные здесь пациенты были гомозиготными по мутации, мы смогли лучше выяснить биохимическое и биологическое воздействие His257.Нормальная мочевая кислота и субстраты PNP, обнаруженные в крови пациентов, предполагают, что PNP сохранял значительную каталитическую активность in vivo . Аналогичным образом, 8–11% ферментативная активность PNP в клетках пациентов II.1 и II.2 согласуется с предыдущим сообщением о 5% каталитической активности после сайт-направленного мутагенеза His257, хотя и с аланином, чем с аспарагиновой кислотой. сообщается здесь (43). Примечательно, что результаты полуколичественного анализа PNP, проведенного здесь, не были нормализованы по содержанию белка, что ограничивает сравнение наших результатов с результатами других лабораторий.Тем не менее, аналогичные значения, полученные с использованием разных типов клеток и здоровых контролей, а также практически идентичное содержание белка PNP при вестерн-блоттинге подтверждают точность анализа активности ферментов, используемого здесь. Взятые вместе, эти результаты показывают, что His257 важен, но незаменим для активности PNP.
В заключение, здесь мы расширяем фенотип наследственных дефектов, связанных с дефицитом PNP, и демонстрируем, что пациенты с частичным дефицитом PNP могут иметь в зрелом возрасте только легкие-умеренные иммунные аномалии и нормальное неврологическое развитие.Мы также показываем, что почти нормальный иммунитет и типичное развитие могут быть достигнуты при 8–11% активности PNP, что предполагает, что такая активность может быть минимумом, необходимым для будущих терапевтических вмешательств.
Заявление о доступности данныхВсе наборы данных, созданные для этого исследования, включены в статью / дополнительный материал.
Заявление об этикеИсследования с участием людей были рассмотрены и одобрены советами по этике исследований Госпитального центра Университета Монреаля, Монреаль, Квебек, и Больницы для больных детей, Торонто, Онтарио.Пациенты / участники предоставили письменное информированное согласие на участие в этом исследовании.
Авторские взносы
EG и HC концептуализировали это исследование и разработали эксперименты. ХК, NC и EG лечили пациентов. Все авторы внесли свой вклад в написание, рецензирование рукописи и визуализацию данных. XX и ML-P проводили эксперименты.
Финансирование
Эта работа была частично поддержана кафедрой иммунологических исследований Кэмпбелла (EG).
Конфликт интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могут быть истолкованы как потенциальный конфликт интересов.
Рецензент MH заявил редактору о прошлом соавторстве с одним из авторов EG.
Благодарности
Авторы благодарят пациентов и их семьи за участие в этом исследовании и постоянное стремление к расширению знаний о дефиците PNP.
Сокращения
ADA, аденозиндезаминаза; AT, Атаксия телеангиэктазии; HSC, гемопоэтические стволовые клетки; КДа, килодатон; Н.К., Натуральный убийца; PBMC, мононуклеарные клетки периферической крови; PNP, пуриновая нуклеозидфосфорилаза; SI — индекс стимуляции; SF — доля выживаемости; ВЗОМТ, первичный иммунодефицит; ТАТПНП, ТАТ, слитый с ПНП.
Список литературы
1. Grunebaum E, Cohen A, Roifman CM. Последние достижения в понимании и устранении дефицита аденозиндезаминазы и пуриновой нуклеозидфосфорилазы. Curr Opin Allergy Clin Immunol . (2013) 13: 630–8. DOI: 10.1097 / ACI.0000000000000006
PubMed Аннотация | CrossRef Полный текст | Google Scholar
2. Арпайя Э., Бенвенисте П., Ди Кристофано А., Гу И, Далал И., Келли С. и др. Митохондриальная основа иммунодефицита. Данные, полученные на мышах с дефицитом пуриновой нуклеозид-фосфорилазы. J Exp Med . (2000) 191: 2197–208. DOI: 10.1084 / jem.191.12.2197
PubMed Аннотация | CrossRef Полный текст | Google Scholar
3. Коэн А., Грюнебаум Э, Арпая Э, Ройфман СМ. Иммунодефицит, вызванный дефицитом пуриновой нуклеозидфосфорилазы. Immunol Allergy Clinics North Am. (2000) 20: 143–59. DOI: 10.1016 / S0889-8561 (05) 70139-9
PubMed Аннотация | CrossRef Полный текст | Google Scholar
4. Дрор Й., Грунебаум Э., Хитцлер Дж., Нарендран А., Йе С., Телье Р. и др.Дефицит пуриновой нуклеозидфосфорилазы, связанный с диспластической морфологией костного мозга. Педиатр Рес . (2004) 55: 472–7. DOI: 10.1203 / 01.PDR.0000111286.23110.F8
PubMed Аннотация | CrossRef Полный текст | Google Scholar
5. Сикпи М.О., Ван И, Малля С.М. Дефектная модуляция репарации двухцепочечных разрывов в клетках с атаксией и телеангиэктазией с помощью гамма-излучения. Радиат Рес . (1998) 150: 627–35. DOI: 10.2307 / 3579885
PubMed Аннотация | CrossRef Полный текст | Google Scholar
6.Гиблетт ER, Амманн А.Дж., Вара Д.В., Сандман Р., Даймонд Л.К. Дефицит нуклеозид-фосфорилазы у ребенка с сильно нарушенным Т-клеточным иммунитетом и нормальным В-клеточным иммунитетом. Ланцет . (1975) 1: 1010–3. DOI: 10.1016 / S0140-6736 (75) 91950-9
PubMed Аннотация | CrossRef Полный текст | Google Scholar
7. Somech R, Lev A, Grisaru-Soen G, Shiran SI, Simon AJ, Grunebaum E. Дефицит пуриновой нуклеозидфосфорилазы, проявляющийся как тяжелый комбинированный иммунодефицит. Иммунол Рес .(2013) 56: 150–4. DOI: 10.1007 / s12026-012-8380-9
PubMed Аннотация | CrossRef Полный текст | Google Scholar
8. Багет С, Вермилен С, Бричард Б., Луи Дж., Дахан К., Винсент М.Ф. и др. Стойкая задержка развития, несмотря на успешную трансплантацию костного мозга по поводу дефицита пуриновой нуклеозидфосфорилазы. J Педиатр Hematol Oncol . (2002) 24: 69–71. DOI: 10.1097 / 00043426-200201000-00018
PubMed Аннотация | CrossRef Полный текст | Google Scholar
9.Мартин Дж., Шарма Р., Нельсон Р.П., Шуберт Ф., Вейда Дж. Первое сообщение о беременности у пациентки с дефицитом пуриновой нуклеозидфосфорилазы. Педиатр плода . (2016) 35: 120–3. DOI: 10.3109 / 15513815.2016.1139020
PubMed Аннотация | CrossRef Полный текст | Google Scholar
10. Рич К.С., Арнольд В.Дж., Палелла Т., Фокс И.Х. Клеточный иммунодефицит с аутоиммунной гемолитической анемией при дефиците пуриновой нуклеозидфосфорилазы. Am J Med . (1979) 67: 172–6.DOI: 10.1016 / 0002-9343 (79) -1
PubMed Аннотация | CrossRef Полный текст | Google Scholar
11. Уотсон А.Р., Эванс Д.И., Марсден Х.Б., Миллер В., Роджерс П.А. Дефицит пуриновой нуклеозидфосфорилазы, связанный со смертельным лимфопролиферативным заболеванием. Арч Дис Детский . (1981) 56: 563–5. DOI: 10.1136 / ADC.56.7.563
PubMed Аннотация | CrossRef Полный текст | Google Scholar
12. Бродски Н., Свенссон М., ван Куйленбург А.Б., Мейер Дж., Зетекоу Л., Труэдссон Л. и др.Новые генетические мутации у первого шведского пациента с дефицитом пуриновой нуклеозидфосфорилазы и клинические исходы после трансплантации гемопоэтических стволовых клеток от HLA-совместимого неродственного донора. Представитель JIMD . (2015) 24: 83–9. DOI: 10.1007 / 8904_2015_444
PubMed Аннотация | CrossRef Полный текст | Google Scholar
13. Мансури А., Мин В., Коул С. Дж., Джосселин С. А., Хендерсон Дж. Т., ван Эде М. и др. Мозжечковые аномалии у мышей с дефицитом пуриновой нуклеозидфосфорилазы. Нейробиол Дис . (2012) 47: 201–9. DOI: 10.1016 / j.nbd.2012.04.001
PubMed Аннотация | CrossRef Полный текст | Google Scholar
14. Парванех Н., Теймуриан С., Джакомелли Дж., Бадалзаде М., Бертелли М., Захарова Э. и др. Новые мутации NP у двух пациентов с дефицитом пуриновой нуклеозидфосфорилазы. Клин Биохим . (2008) 41: 350–2. DOI: 10.1016 / j.clinbiochem.2007.11.007
PubMed Аннотация | CrossRef Полный текст | Google Scholar
15.Айтекин С., Догу Ф., Танир Г., Гулоглу Д., Сантистебан И., Хершфилд М.С. и др. Дефицит пуриновой нуклеозидфосфорилазы со смертельным исходом у двух сестер. Eur J Pediatr . (2010) 169: 311–4. DOI: 10.1007 / s00431-009-1029-6
PubMed Аннотация | CrossRef Полный текст | Google Scholar
16. Шах Н., Лингаппа Л., Конанки Р., Рани С., Ведам Р., Муруган С. Иммунодефицит, моторная задержка и гипоурикемия, вызванные новой мутацией гена пуриновой нуклеозидфосфорилазы у индийского младенца. Энн Индийский Академик Нейрол . (2019) 22: 231–3. DOI: 10.4103 / aian.AIAN_430_17
PubMed Аннотация | CrossRef Полный текст | Google Scholar
17. Брум С.Б., Грэм М.Л., Солсбери Ф.Т., Хершфилд М.С., Бакли Р.Х. Коррекция дефицита пуриновой нуклеозидфосфорилазы путем трансплантации аллогенного костного мозга от брата или сестры. Дж. Педиатр . (1996) 128: 373–6. DOI: 10.1016 / S0022-3476 (96) 70285-8
PubMed Аннотация | CrossRef Полный текст | Google Scholar
18.Карпентер PA, Ziegler JB, Vowels MR. Поздняя диагностика и коррекция недостаточности пуриновой нуклеозидфосфорилазы с помощью аллогенной трансплантации костного мозга. Пересадка костного мозга . (1996) 17: 121–4.
PubMed Аннотация | Google Scholar
19. Майерс Л.А., Хершфилд М.С., Нил В.Т., Эсколар М., Курцберг Дж. Дефицит пуриновой нуклеозидфосфорилазы, проявляющийся лимфопенией и задержкой развития: успешная коррекция с помощью трансплантации пуповинной крови. Дж. Педиатр . (2004) 145: 710–2. DOI: 10.1016 / j.jpeds.2004.06.075
PubMed Аннотация | CrossRef Полный текст | Google Scholar
20. Delicou S, Kitra-Roussou V, Peristeri J, Goussetis E, Vessalas G, Rigatou E, et al. Успешная трансплантация HLA-идентичных гемопоэтических стволовых клеток пациенту с дефицитом пуриновой нуклеозидфосфорилазы. Педиатр по трансплантологии . (2007) 11: 799–803. DOI: 10.1111 / j.1399-3046.2007.00772.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
21.Йейтс Л., Слаттер М.А., Геннери А.Р. Инфузия костного мозга родственного брата пациенту с дефицитом пуриновой нуклеозидфосфорилазы приводит к расщеплению смешанного донорского химеризма и нормальному иммунитету. Передний педиатр . (2017) 5: 143. DOI: 10.3389 / fped.2017.00143
CrossRef Полный текст | Google Scholar
22. Хениг М., Альберт М.Х., Шульц А., Спарбер-Зауэр М., Шютц С., Белоградский Б. и др. Пациенты с дефицитом аденозиндезаминазы, выжившие после трансплантации гемопоэтических стволовых клеток, имеют высокий риск осложнений со стороны ЦНС. Кровь . (2007) 109: 3595–602. DOI: 10.1182 / кровь-2006-07-034678
PubMed Аннотация | CrossRef Полный текст | Google Scholar
24. Торо А., Грунебаум Э. ТАТ-опосредованная внутриклеточная доставка пуриновой нуклеозидфосфорилазы исправляет ее дефицит у мышей. Дж. Клин Инвест . (2006) 116: 2717–26. DOI: 10.1172 / JCI25052
PubMed Аннотация | CrossRef Полный текст | Google Scholar
25. Ляо П, Торо А., Мин В., Ли С., Ройфман К. М., Грюнебаум Э.Лентивирусная генная терапия недостаточности пуриновой нуклеозидфосфорилазы. Дж. Джин Мед . (2008) 10: 1282–93. DOI: 10.1002 / JGM.1261
PubMed Аннотация | CrossRef Полный текст | Google Scholar
26. Грюнебаум Э., Маццолари Э., Порта Ф., Даллера Д., Аткинсон А., Рид Б. и др. Трансплантация костного мозга при тяжелом комбинированном иммунодефиците. ДЖАМА . (2006) 295: 508–18. DOI: 10.1001 / jama.295.5.508
PubMed Аннотация | CrossRef Полный текст | Google Scholar
27.Райксен Г., Куис В., Вадман С.К., Спапен Л.Дж., Дюран М., Воорбруд Б.С. и др. Новый случай недостаточности пуриновой нуклеозидфосфорилазы: энзимологические, клинические и иммунологические характеристики. Педиатр Рес . (1987) 21: 137–41. DOI: 10.1203 / 00006450-198702000-00006
PubMed Аннотация | CrossRef Полный текст | Google Scholar
28. Wroblewski JM, Copple A, Batson LP, Landers CD, Yannelli JR. Фенотипирование клеточной поверхности и продукция цитокинов линиями лимфобластоидных клеток (LCL), трансформированными вирусом Эпштейна-Барра (EBV). Дж. Иммунол Методы . (2002) 264: 19–28. DOI: 10.1016 / S0022-1759 (01) 00565-8
PubMed Аннотация | CrossRef Полный текст | Google Scholar
29. Аль-Дирбаши О.Ю., Огрел С., МакИнтош Н., Хиггинс Л., Мак-Робертс С., Фишер Л. и др. Тандемное масс-спектрометрическое определение пуриновых метаболитов и активности аденозиндезаминазы для скрининга новорожденных на ADA – SCID. LymphoSign J. (2015) 2: 135–45. DOI: 10.14785 / lpsn-2014-0024
CrossRef Полный текст | Google Scholar
30.Торо А., Пайва М., Акерли С., Грунебаум Е. Внутриклеточная доставка пуриновой нуклеозидфосфорилазы (PNP), слитой с доменом трансдукции белка, корректирует дефицит PNP in vitro . Клеточный иммунол . (2006) 240: 107–15. DOI: 10.1016 / j.cellimm.2006.07.003
PubMed Аннотация | CrossRef Полный текст | Google Scholar
31. Далал И., Грюнебаум Э, Коэн А., Ройфман СМ. Две новые мутации у пациента с дефицитом пуриновой нуклеозид фосфорилазы (PNP). Клин Генет .(2001) 59: 430–7. DOI: 10.1034 / j.1399-0004.2001.5.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
32. Сунь Х, Беккер-Катания С.Г., Чун Х.Х., Хван М.Дж., Хо Й., Ван З. и др. Ранняя диагностика атаксии и телеангиэктазии с помощью теста на радиочувствительность. Дж. Педиатр . (2002) 140: 724–31. DOI: 10.1067 / mpd.2002.123879
PubMed Аннотация | CrossRef Полный текст | Google Scholar
33. Walker PL, Corrigan A, Arenas M, Escuredo E, Fairbanks L, Marinaki A.Дефицит пуриновой нуклеозидфосфорилазы: обновление мутации. Нуклеотидные нуклеотидные кислоты . (2011) 30: 1243–7. DOI: 10.1080 / 15257770.2011.630852
PubMed Аннотация | CrossRef Полный текст | Google Scholar
34. Йенч-Ульрих К., Кенигсманн М., Морен М., Франке А. Референсные диапазоны субпопуляций лимфоцитов в сбалансированной по возрасту и полу популяции из 100 здоровых взрослых — моноцентрическое немецкое исследование. Клин Иммунол . (2005) 116: 192–7. DOI: 10.1016 / j.clim.2005.03.020
PubMed Аннотация | CrossRef Полный текст | Google Scholar
35. Фекрванд С., Яздани Р., Аболхассани Х., Гаффари Дж., Агамохаммади А. Первый пациент с дефицитом пуриновой нуклеозидфосфорилазы, напоминающий дефицит IgA, и обзор литературы. Иммунол Инвест . (2019) 48: 410–30. DOI: 10.1080 / 08820139.2019.1570249
PubMed Аннотация | CrossRef Полный текст | Google Scholar
36. Селмели Ф, Тюрккахраман Д, Уйгун В, ла Марка Г, Хершфилд М, Есилипек А.Успешная трансплантация неродственных стволовых клеток периферической крови с режимом кондиционирования с пониженной интенсивностью у пациента с поздним началом дефицита пуриновой нуклеозидфосфорилазы. Педиатр по трансплантологии . (2015) 19: E47–50. DOI: 10.1111 / petr.12413
CrossRef Полный текст | Google Scholar
37. Тиммс PM, Симмондс HA, Bold AM. Заметная гипоурикемия при дефиците пуриновой нуклеозидфосфорилазы — случайное обнаружение на экране. Clin Chem . (1996) 42 (6, ч. 1): 985.DOI: 10.1093 / Clinchem / 42.6.985a
PubMed Аннотация | CrossRef Полный текст | Google Scholar
38. Corneo B, Moshous D, Güngör T, Wulffraat N, Philippet P, Le Deist FL, et al. Идентичные мутации в генах RAG1 или RAG2, ведущие к дефектной активности рекомбиназы V (D) J, могут вызывать либо тяжелый комбинированный иммунодефицит T-B, либо синдром Оменна. Кровь . (2001) 97: 2772–6. DOI: 10.1182 / blood.V97.9.2772
CrossRef Полный текст | Google Scholar
39.Yu Y, Arora A, Min W., Roifman CM, Grunebaum E. Включение EdU представляет собой альтернативный нерадиоактивный анализ захвата [(3) H] тимидина для измерения in vitro пролиферации Т-клеток мышей. Дж. Иммунол Методы . (2009) 350: 29–35. DOI: 10.1016 / j.jim.2009.07.008
PubMed Аннотация | CrossRef Полный текст | Google Scholar
40. Маркерт М.Л., Хершфилд М.С., Шифф Р.И., Бакли Р.Х. Дефицит аденозиндезаминазы и пуриннуклеозидфосфорилазы: оценка терапевтических вмешательств у восьми пациентов. Дж. Клин Иммунол . (1987) 7: 389–99. DOI: 10.1007 / BF00917017
PubMed Аннотация | CrossRef Полный текст | Google Scholar
41. Хо М.К., Ши В., Ринальдо-Маттис А., Тайлер П.С., Эванс Г.Б., Клинч К. и др. Четыре поколения аналогов пуриновой нуклеозид фосфорилазы в переходном состоянии человека. Proc Natl Acad Sci USA . (2010) 107: 4805–12. DOI: 10.1073 / pnas.09107
PubMed Аннотация | CrossRef Полный текст | Google Scholar
42. Фурман Р.Р., Хельцер Д.Ингибирование пуриновой нуклеозидфосфорилазы как новый терапевтический подход к В-клеточным лимфоидным злокачественным новообразованиям. Семин Онкол . (2007) 34 (6 доп.). 5: S29–34. DOI: 10.1053 / j.seminoncol.2007.11.004
PubMed Аннотация | CrossRef Полный текст | Google Scholar
43. Erion MD, Takabayashi K, Smith HB, Kessi J, Wagner S, Hönger S и др. Пуриновая нуклеозидфосфорилаза. 1. Структурно-функциональные исследования. Биохимия . (1997) 36: 11725–34. DOI: 10.1021 / bi961969w
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Предварительная замена ферментов путем переливания эритроцитов при дефиците PNP
Дефицит пуриннуклеозидфосфорилазы (PNP) — редкое нарушение обмена веществ, приводящее к комбинированному иммунодефициту (CID) и неврологическим нарушениям, таким как задержка развития, атаксия и двигательные нарушения.PNP сильно экспрессируется в красных кровяных тельцах (RBC) и фосфоролизирует (дезокси) -инозин и (дезокси) -гуанозин до (дезокси) -гуанина и гипоксантина. Из-за ферментативного блока у пациентов с дефицитом PNP наблюдаются повышенные концентрации пуриновых (дезокси) нуклеозидов, которые обладают лимфо- и нейротоксическим действием. Например, dGTP, который накапливается в митохондриях, подавляет синтез митохондриальной ДНК, что приводит к апоптозу и способствует неврологическому дефициту [1]. Метаболический блок, вызванный дефицитом PNP, предотвращает образование мочевой кислоты и часто приводит к гипоурикемии [2].Дефицит PNP можно лечить с помощью HSCT, что приводит к коррекции CID, но неврологический дефицит часто сохраняется или прогрессирует. Новаторская работа по проверке концепции, проведенная в 1980-х годах, показала, что переливание эритроцитов улучшает метаболические и иммунологические функции [3, 4]. С внедрением скрининга новорожденных на основе TREC и возможностью скрининга дефицита PNP с помощью тандемной масс-спектрометрии, которая будет более чувствительной, растет потребность в ведении бессимптомных новорожденных пациентов с PNP (P), не только излечивая CID, но и предотвращая неврологический дефицит [5].
Мы сообщаем о серии из трех новорожденных / младенцев, которым был поставлен генетический диагноз дефицита PNP пренатально (P1 и P2, братья и сестры) или в раннем возрасте (P3) из-за семейного индекса (таблица S1). Все они получали последовательную терапию с немедленным и постоянным переливанием эритроцитов (ЭТ) с последующей ТГСК (рис. 1). Перед первоначальным обменным переливанием эритроцитов (EET) активность фермента PNP RBC была снижена до 4,8 ± 3,3 мкмоль / мин / г гемоглобина (эталон 60–100), а метаболиты пурина в сухих пятнах крови были повышены (e.г., гуанозин 12,3 ± 5,5 мкмоль / л; ссылка <1.1, рис. 1, таб.S3 – S5). Уровень мочевой кислоты был снижен только для P3 (0,2 мг / дл мочевой кислоты на 6-й день жизни), тогда как он был нормальным или слегка повышенным для P1 и P2 (рис. S2). В то время как послеродовое клиническое обследование и ультразвуковое исследование головного мозга (пациенты 1 и 2) не выявили неврологических отклонений, наблюдалась разная степень снижения иммунных клеток (Таблица S2). Первоначальный EET был выполнен сразу после рождения в P1 и P2, и на 23-й день жизни в P3. Эти обменные переливания эритроцитов на 1-й день жизни привели к увеличению активности фермента PNP до 50 (P1) и 21 (P2) мкмоль / мин / г гемоглобина (на d2 жизни) и нормализации метаболитов пурина (на примере гуанозина). 0.5 ± 0,1 мкмоль / л) (рис. 1, таб.S4 – S6). После этого P1 – P3 регулярно получали ET / EET по индивидуальным схемам, которые руководствовались пуриновыми метаболитами (рис. 1, таб.S4 – S6). P1 получал пять дополнительных ЭТ каждые 11 ± 10 дней в объеме 15 ± 9 мл / кг без предварительной флеботомии из-за анемии, вызванной сопутствующей серповидно-клеточной анемией (рис. 1а и таблица S4). P2 получил девять дополнительных EET каждые 12 ± 5 дней в объеме 14 ± 2 мл / кг, которым предшествовали 6 кровопусканий по 10 ± 4 мл / кг (рис. 1b и таблица S5). P3 получал 2 дополнительных ЭТ каждые 24 ± 6 дней в объеме 24 мл / кг (43-й день) и неясном объеме ET (71-й день) (рис.1c и Табл.S6). После первоначального EET у P1 и P2 была тромбоцитопения (41 г / л и 28 г / л) без признаков кровотечения, и обоим переливали тромбоциты один раз. P1 имел преходящую частичную респираторную недостаточность с кислородной зависимостью через носовую канюлю, а P2 имел небольшую перегрузку жидкостью и получил две дозы фуросемида. У P3 была легкая тромбоцитопения после первого (108 г / л) и после второго ЭТ (138 г / л). Других побочных эффектов, связанных с EET / ET, не наблюдалось.
Рис.1Клинический курс пациентов 1 ( a ), 2 ( b ) и 3 ( c ) с указанием временных точек переливаний эритроцитов (EET / ETs) и HSCT, соответствующих уровней гуанозина из пятен засохшей крови (черные точки) и химеризма цельной крови после ТГСК (серая, штрихпунктирная линия, треугольники).Отображаются только ЭТ до ТГСК, а не во время аплазии после химиотерапии. Верхнее контрольное значение для гуанозина (1,1 мкмоль / л) отмечено черной пунктирной линией. ЦСЖ, спинномозговая жидкость
Для достижения окончательного излечения от дефицита PNP, P1 – P3 подверглись аллогенной ТГСК после миелоаблативного кондиционирования на основе бусульфана / флударабина / алемтузумаба из несоответствующей семьи или неродственных доноров в возрасте от 64 до 117 дней соответственно (Табл. S1). ТГСК P1 осложнилась веноокклюзионной болезнью, которая ответила на дефибротид, и вторичной недостаточностью трансплантата (рис.1, таб.S1 и S4). В то время как химеризм доноров периферической крови снижался (72% в день +196), токсичные метаболиты пурина увеличивались в периферической крови и спинномозговой жидкости. В этот момент времени у P1 не было признаков CID, но неврологическое развитие замедлилось. Между постановкой диагноза вторичной недостаточности трансплантата и повторной ТГСК ЭТ не проводились. Пациенту 1 без осложнений была проведена повторная трансплантация в возрасте 19 месяцев от того же донора с альтернативным режимом миелоабляции и источником стволовых клеток (Табл.S1). P2 пережил острую болезнь «трансплантат против хозяина» (GVHD) ° III, которая отреагировала на иммуносупрессивную терапию. Трансплантация P3 прошла без осложнений. При последующем наблюдении 42, 32 и 37 месяцев, соответственно, все пациенты живы и здоровы с полным восстановлением иммунитета и отсутствием хронической РТПХ.
P1 – P3 последовательно достигли этапов развития при последующих визитах; однако у них наблюдается глобальная задержка в развитии разной степени (Табл. S3). P1 и P3 смогли частично догнать когнитивное развитие, в то время как P2 показал более медленный прогресс.Анализируя уровни гуанозина с течением времени (рис. 1), трудно определить различные AUC. Более того, множество дополнительных факторов — например, разные генотипы (P1 / 2 по сравнению с P3), внутриутробное воздействие токсичных пуриновых метаболитов [6], 2-я ТГСК в P1, длительное употребление глюкокортикоидов и неспособность развиваться из-за острой GVHD в P2 — различают P1 – P3 и могут влиять на неврологическое развитие. В частности, время воздействия существенно повышенных нейротоксических метаболитов может влиять на двигательное развитие.P3, которая начала ET / EETs в возрасте 23 дней и, таким образом, провела большую часть своего новорожденного периода с высокотоксичными уровнями, не может ходить самостоятельно в возрасте 3 лет, в то время как P1 и P2 подвижны сами по себе. Однако развитие моторики наиболее нарушено в P1 – P3, и P3 лучше справляется с другими областями тестирования по сравнению с P1 и P2. Нейровизуализация P3 выявляет глобальную атрофию головного мозга (рис. S1) в возрасте 28 месяцев, в то время как для P1 и P2 нет.
Наблюдения за пациентами с одиночным ПНП показывают, что неврологический дефицит может улучшиться после ранней ТГСК, но также может прогрессировать, особенно после поздней ТГСК [7, 8].Очевидно, что отсутствуют более крупные серии ТГСК с данными по долгосрочным исходам. Более того, в ранее существовавшей литературе не было показано четких корреляций генотип-фенотип для неврологического исхода, и на развитие детей также влияют приобретенные инфекции [9]. Таким образом, невозможно оценить неврологический исход у одного пациента и сложно оценить эффект терапевтических вмешательств. Нелеченый дефицит мышиного ПНП вызывает сопоставимые неврологические нарушения, которые можно предотвратить с помощью ФЗТ [10].Кратковременное лечение ЭТ для обеспечения фермента PNP доказало принципиальное доказательство того, что это может привести к краткосрочному биохимическому и иммунологическому улучшению 34 . Здесь мы демонстрируем на серии случаев новорожденных / младенцев с дефицитом PNP, что этот подход безопасен в качестве предварительной стратегии с последующей ранней окончательной ТГСК у пациентов с предсимптоматическими симптомами. Как только состояние новорожденного станет стабильным, начальное EET должно проводиться под тщательным наблюдением опытной команды в известном экспертном центре, и особое внимание следует уделять гипокальциемии, гипомагниемии и тромбоцитопении [11, 12].Обнаружение метаболитов пурина с помощью тандемной масс-спектрометрии позволяет управлять режимами ЭТ и оптимизировать количество переливаний эритроцитов, тем самым поддерживая метаболиты пурина на более низком уровне и избегая ненужной перегрузки железом, вызванной переливанием крови. Уровни мочевой кислоты у наших трех пациентов недостаточно хорошо соответствовали уровням метаболитов пурина и не могли использоваться для руководства переливанием (рис. S2), что оправдывает более сложное применение тандемной масс-спектрометрии для прямого измерения метаболитов пурина.Мы показываем, что этот подход приводит к коррекции метаболитов пуринов и стабилизирует иммунологические фенотипы (Табл.S7 – S8). Мы предполагаем, что улучшенной нейрозащиты можно достичь немедленной (EET), последовательной ET и длительной перекрестной детоксикацией (HSCT), даже несмотря на то, что значительный долгосрочный неврологический эффект еще предстоит определить. Мы предполагаем, что возможная неврологическая польза перевешивает возможные осложнения EET / ET, которые могут быть применены в качестве моста к HSCT в кратчайшие сроки.Мы предполагаем, что внутриутробные ETs у плодов с дефицитом PNP могут еще больше улучшить их нейрокогнитивное развитие.
При незлокачественных иммунологических и метаболических нарушениях степень донорского химеризма, необходимая для лечения, значительно варьируется в зависимости от заболеваний и конкретных клеточных линий [13]. При дефиците PNP потребность центральной нервной системы в активности фермента PNP может быть нацелена только косвенно с помощью HSCT. Таким образом, режим кондиционирования должен обеспечивать миграцию донорских моноцитов в центральную нервную систему и последующую трансдифференцировку в микроглиеподобные клетки.На животных моделях этого лучше всего можно было достичь с помощью кондиционирования, содержащего бусульфан, которое мы выбрали для P1 – P3. Следовательно, необходимо получить высокий донорский миелоидный химеризм и надлежащим образом контролировать его. Хотя эритроциты представляют собой основной источник PNP, их химеризм нелегко оценить в периферической крови с помощью стандартных генетических методов. Мы показываем, что мониторинг пуриновых метаболитов может обнаруживать повышение метаболитов как следствие неполного эритроцита и миелоидного химеризма, и предлагаем его в качестве метода мониторинга дефицита ADA и PNP после HSCT и генной терапии.
Таким образом, терапия дефицита PNP с предварительным замещением ферментов посредством EET / ET в сочетании с окончательной ТГСК приводит к немедленной и постоянной коррекции метаболических и иммунологических фенотипов, в то время как долгосрочные неврологические преимущества еще предстоит определить. Мониторинг пуриновых метаболитов с помощью тандемной масс-спектрометрии возможен для управления отдельными режимами ЭТ и для обнаружения ранней недостаточности трансплантата, особенно эритроидного происхождения.
Доступность данных
Дополнительные данные — в дополнительном материале.
Ссылки
- 1.
Arpaia E, Benveniste P, di Cristofano A, Gu Y, Dalal I, Kelly S, et al. Митохондриальная основа иммунодефицита. Данные, полученные на мышах с дефицитом пуриновой нуклеозид-фосфорилазы. J Exp Med . 2000; 191: 2197–208. https://doi.org/10.1084/jem.191.12.2197.
CAS Статья PubMed PubMed Central Google Scholar
- 2.
Ивахана Х., Итакура М. Наследственные нарушения метаболизма мочевой кислоты — классификация, ферментативная и ДНК-диагностика.Нихон Риншо. 1996; 54: 3303–8.
CAS PubMed Google Scholar
- 3.
Rich KC, Majias E, Fox IH. Дефицит пуриновой нуклеозидфосфорилазы: улучшение метаболических и иммунологических функций при переливании эритроцитов. N Engl J Med. 1980; 303: 973–7. https://doi.org/10.1056/NEJM198010233031705.
CAS Статья PubMed Google Scholar
- 4.
Стаал Г.Е., Ступ Дж. В., Зегерс Б. Дж., Зигенбек ван Хеукелом Л. Х., ван дер Влист М. Дж., Вадман С. К. и др.Метаболизм эритроцитов при дефиците пуриновой нуклеозидфосфорилазы после заместительной ферментной терапии инфузией эритроцитов. J Clin Invest. 1980; 65: 103–8. https://doi.org/10.1172/JCI109639.
CAS Статья PubMed PubMed Central Google Scholar
- 5.
la Marca G, Canessa C, Giocaliere E, Romano F, Malvagia S, Funghini S, et al. Диагностика иммунодефицита, вызванного дефектом пуриновой нуклеозидфосфорилазы, с помощью тандемной масс-спектрометрии на сухих пятнах крови.J Allergy Clin Immunol. 2014; 134: 155–9. https://doi.org/10.1016/j.jaci.2014.01.040.
CAS Статья PubMed Google Scholar
- 6.
Kleijer WJ, Hussaarts-Odijk LM, Los FJ, Pijpers L, de Bree PK, Duran M. Пренатальная диагностика дефицита пуриновой нуклеозидфосфорилазы в первом и втором триместрах беременности. Prenat Diagn. 1989; 9: 401–7. https://doi.org/10.1002/pd.19700.
CAS Статья PubMed Google Scholar
- 7.
Сингх В. Перекрестная коррекция недостаточности пуриновых нуклеозидфосфорилазы после трансплантации гемопоэтических стволовых клеток: привитые донорские лейкоциты обеспечивают фермент остаточными реципиентными клетками с дефицитом фермента. JIMD Rep. 2012; 6: 39–42. https://doi.org/10.1007/8904_2012_126.
Артикул PubMed PubMed Central Google Scholar
- 8.
Майерс Л.А., Хершфилд М.С., Нил В.Т., Эсколар М., Курцберг Дж. Дефицит пуриновой нуклеозидфосфорилазы (PNP-def), проявляющийся лимфопенией и задержкой развития: успешная коррекция с помощью трансплантации пуповинной крови.J Pediatr. 2004. 145: 710–2. https://doi.org/10.1016/j.jpeds.2004.06.075.
CAS Статья PubMed Google Scholar
- 9.
Schejter YD, Even-Or E, Shadur B, NaserEddin A, Stepensky P, Zaidman I. Широкий клинический спектр и результаты трансплантации дефицита PNP. J Clin Immunol. 2020; 40: 123–30. https://doi.org/10.1007/s10875-019-00698-1.
Артикул PubMed Google Scholar
- 10.
Мансури А., Мин В., Коул С.Дж., Джосселин С.А., Хендерсон Дж.Т., ван Эеде М. и др. Мозжечковые аномалии у мышей с дефицитом пуриновой нуклеозидфосфорилазы. Neurobiol Dis. 2012; 47: 201–9. https://doi.org/10.1016/j.nbd.2012.04.001.
CAS Статья PubMed Google Scholar
- 11.
Boskabadi HMM. Оценка степени тяжести и продолжительности тромбоцитопении после обменного переливания крови при неонатальной гипербилирубинемии. Иран Дж. Пед Гематол Онкол .2019; 9 : 91–7. https://doi.org/10.18502/ijpho.v9i2.608.
Артикул Google Scholar
- 12.
Chcacham SKJ, Dutta S, Kumar P. Неблагоприятные события после переливания крови при неонатальной гипербилирубинемии: проспективное исследование. Журнал клинической неонатологии. 2019; 8: 79–84.
Артикул Google Scholar
- 13.
Фитцхью С.Д., Кордес С., Тейлор Т., Коулз В., Роском К., Линк М. и др.По крайней мере, 20% донорского миелоидного химеризма необходимо для изменения серповидного фенотипа после аллогенной ТГСК. Кровь. 2017; 130: 1946–8. https://doi.org/10.1182/blood-2017-03-772392.
CAS Статья PubMed PubMed Central Google Scholar
Скачать ссылки
Благодарности
Мы ценим личный вклад пациентов и их семей. Мы хотели бы поблагодарить медсестер и медицинские бригады отделения детской иммунологии и гематоонкологии, особенно Анн-Мадлен Бёкстегерс, Ситу Хейде, Мари-Кристин Колокитас и Лизу-Мари Кёлер из LMU в Мюнхене, а также Йоханнеса Шульте и Уве Кёльша из Charité Berlin.Мы благодарим Еву Эйсл, Маюми Хофманн и Ирмгард Эккерляйн за техническую помощь в Лаборатории иммунологической диагностики детской больницы доктора фон Хаунера и Сабрину Мальваджа за измерение метаболитов пурина во Флорентийском университете. Мы благодарим нейропедиатрические бригады LMU Munich и Charité Berlin, а также радиологическое отделение Charité Berlin. Кристоф Кляйн и Раффаэле Конка оказывали постоянную поддержку и плодотворные обсуждения.
Код доступности
Не применимо.
Финансирование
Финансирование в открытом доступе, организованное Projekt DEAL. FH получил финансирование от Фонда Care-for-Rare (C4R, 160073), Немецкого центра исследований инфекций (DZIF, TTU 07.909), Else Kröner-Fresenius Stiftung (EKFS, 2017_A110) и Федерального министерства образования и Германии Германии. Исследования (BMBF, 01GM1910C). C.K., M.H.A. и F.H. являются членами Европейской справочной сети по редким иммунодефицитным, аутовоспалительным и аутоиммунным заболеваниям — Project ID No.739543.
Информация об авторе
Заметки автораАнна Эйхингер, Хорст фон Бернут, Майкл Х. Альберт и Фабиан Хаук внесли равный вклад в эту работу.
Филиалы
Отделение педиатрии, Детская больница доктора фон Хаунера, Университетская больница, Университет Людвига-Максимилиана, Мюнхен, Мюнхен, Германия
Анна Эйхингер, Себастьян А. Шредер, Майкл Х. Альберт и Фабиан Хаунер
Отделение детской пульмонологии, иммунологии и интенсивной терапии, Charité — Universitätsmedizin Berlin, Берлин, Германия
Horst von Bernuth & Cinzia Dedieu
Берлин-Бранденбург Центр регенеративной терапии в Германии
, Берлин в БерлинеОтделение иммунологии, Labor Berlin GmbH, Берлин, Германия
Хорст фон Бернут
Отделение детской гематологии и онкологии, Charité — Universitätsmedizin Berlin, Берлин, Германия
Cinzia Screening
- u Newborn Лаборатория биохимии и фармакологии, Детская клиника N еврология, Детская больница Университета Мейера, Флоренция, Италия
Джанкарло ла Марка
Кафедра экспериментальных и клинических биомедицинских наук, Университет Флоренции, Флоренция, Италия
Джанкарло ла Марка
Немецкий центр исследований инфекций (DZIF ), Мюнхен, Германия
Фабиан Хаук
Мюнхенский центр редких заболеваний (MZSE), Мюнхен, Германия
Фабиан Хаук
Взносы
M.H.A., H.v.B. и F.H. разработали терапевтический режим. G.l.M. проведен метаболический анализ. A.E., C.D., S.A.S, M.H.A., H.v.B. и F.H. лечили пациентов и анализировали данные. A.E., M.H.A. и F.H. написали рукопись. Все авторы предоставили критические отзывы и помогли сформировать исследование, анализ и рукопись.
Автор для переписки
Переписка на Фабиан Хаук.
Декларации этики
Сертификат этики
Рукопись не содержит исследований на людях, но содержит данные ретроспективного обзора диаграмм.Таким образом, рукопись не подлежала утверждению местным этическим комитетом.
Согласие на участие
Было получено информированное согласие всех участвующих лиц, осуществляющих уход.
Согласие на публикацию
Было получено информированное согласие всех участвующих лиц, осуществляющих уход.
Конфликт интересов
Авторы заявляют об отсутствии конкурирующих интересов.
Дополнительная информация
Примечание издателя
Springer Nature сохраняет нейтралитет в отношении юрисдикционных претензий на опубликованных картах и принадлежностей организаций.
Дополнительная информация
Права и разрешения
Открытый доступ Эта статья находится под лицензией Creative Commons Attribution 4.0 International License, которая разрешает использование, совместное использование, адаптацию, распространение и воспроизведение на любом носителе или любом формате, при условии, что вы укажете соответствующую ссылку на оригинального автора (авторов) и источник, укажите ссылку на лицензию Creative Commons и укажите, были ли внесены изменения.Изображения или другие сторонние материалы в этой статье включены в лицензию Creative Commons для статьи, если иное не указано в кредитной линии для материала. Если материал не включен в лицензию Creative Commons для статьи и ваше предполагаемое использование не разрешено законом или превышает разрешенное использование, вам необходимо получить разрешение непосредственно от правообладателя. Чтобы просмотреть копию этой лицензии, посетите http://creativecommons.org/licenses/by/4.0/.
Перепечатки и разрешения
Об этой статье
Цитируйте эту статью
Eichinger, A., von Bernuth, H., Dedieu, C. et al. Предварительная замена ферментов путем переливания эритроцитов при дефиците PNP. J Clin Immunol 41, 1112–1115 (2021). https://doi.org/10.1007/s10875-021-01003-9
Скачать цитату
Успешная трансплантация HLA-идентичного костного мозга пациенту с дефицитом PNP с использованием бусульфана и флударабина для кондиционирования
История болезни
Женщина пациентка родилась третьим ребенком от здоровых, неродственных родителей; в семье не было наследственных болезней или ранних смертей.Беременность протекала без осложнений, но роды в срок (4250 г, 54 см) осложнились дистокией правого плеча, что привело к преходящему парезу верхнего сплетения. С шестого месяца жизни стала очевидной мышечная гипотония туловища. В возрасте 1 года пациентка могла поворачиваться, но не могла ни сидеть, ни ползать. Когда ее приводили в сидячее положение, она имела тенденцию падать на бок или назад. Обнаружена легкая атаксия рук и ног с легкой спастической мышечной гипертонией. Спонтанные движения уменьшились.Не было явных нарушений психосоциального развития или интереса к ее окружению. Пациентка прошла все регулярные прививки, включая живую вакцинацию против БЦЖ и полиомиелита, без осложнений. В возрасте 22 месяцев у девочки не было опасных для жизни инфекций. Однако инфекция ветряной оспы в возрасте 20 месяцев была относительно длительной, несмотря на введение ацикловира.
Лабораторные исследования показали легкую анемию с гемоглобином 97 г / л и лимфопению (500 / мкл).В остальном анализ крови был нормальным, как и электролиты, креатинин, АМК, АСТ, АЛТ, γGT, ЛДГ, липаза, амилаза, параметры свертывания крови, витаминный статус и статус гормонов щитовидной железы. Уровень мочевой кислоты в плазме был очень низким — 3 мкмоль / л. Скрининг на антенатальные инфекции (TORCH) был отрицательным. Аминокислоты и органические кислоты в моче в норме. В спинномозговой жидкости белок, глюкоза, лактат, нейромедиаторы и аминокислоты, включая глицин, были нормальными. Экскреция мочевой кислоты с мочой была очень низкой (43.9 ммоль / моль креатинина) с нормальными значениями ксантина и гипоксантина. Инозин, гуанозин, 2′-дезоксигуанозин и 2′-дезоксиинозин в моче были сильно повышены, что указывает на дефицит PNP. Активность ПНФ в эритроцитах была равна нулю (табл. 1). Таким образом, был подтвержден абсолютный дефицит PNP. Оба родителя и двое братьев и сестер пациента были гетерозиготными по заболеванию (таблица 1). Иммунологические исследования показали тяжелый Т-клеточный иммунодефицит у ребенка (Таблица 2) с нормальным уровнем иммуноглобулинов в сыворотке крови на момент обращения.
Таблица 1 Значения PNP (нмоль / мг Hb / ч) (диапазон контроля 3000–7000) Таблица 2a Иммунологические исследования: распределение субпопуляций лимфоцитов (процентное и абсолютное количество на мкл)Нейрофизиологические исследования, включая ЭЭГ, скорость нервной проводимости и краниоспинальная магнитно-резонансная томография были нормальными. Офтальмологический статус в норме. При ультразвуковом исследовании вилочковая железа выглядела неоднородной со множеством неровностей на УЗИ, а загрудинная часть вилочковой железы была очень тонкой (размер: 31 × 11 × 29 мм).Ультразвуковое исследование всех остальных органов в норме. Другие технические обследования, которые включали кардиологическое обследование, рентген грудной клетки и ЭЭГ, были нормальными.
Старший брат оказался HLA-идентичным, и пациент был направлен на ТКМ в возрасте 25 месяцев.
Состояние до BMT
При поступлении пациент был в хорошем клиническом состоянии, без инфекций, лимфатических узлов нормального размера и видимых миндалин. Отмечалась выраженная мышечная гипотония туловища с кифозом и гипертонией и гиперрефлексией конечностей; можно было сидеть, но не было ни ползания, ни ходьбы.Пациент говорил только отдельные слова, но, казалось, понимал целые предложения. После получения полного информированного согласия обоих родителей ТКМ была проведена с использованием экспериментального режима кондиционирования с бусульфаном и флударабином.
Трансплантация костного мозга
Кондиционирование перед BMT включало бусульфан 5 мг / кг перорально. в четыре приема в день в течение 4 дней (день -13 — день -10; общая доза 20 мг / кг) и флударабин 40 мг / м 2 в виде одной дозы ежедневно в течение 5 дней (день -8 — день -4; всего доза 200 мг / м 2 ).В день 0 из 5-летнего HLA-идентичного брата, совместимого с группой крови, инфузировали 420 мл костного мозга, содержащего 5,7 × 10 8 ядерных клеток на кг. Профилактика GVHD состояла из циклоспорина A, начиная с дня -1, с уровнями в сыворотке от 50 до 100 мкг / л, которые снижались с дня +35 до дня +49. Токсичность от кондиционирования выражалась в облысении и мукозите (1-й степени). Гематологическое восстановление произошло в подходящее время с нейтрофилами> 1000 / мкл на +26 день и ретикулоцитами> 20% на +45 день.Переливание тромбоцитов потребовалось только один раз (день +6), и после этого тромбоциты оставались стабильными> 20 000 / мкл. Приживление было подтверждено на +95 день типированием группы крови и идентификацией нормальной активности PNP в мононуклеарных клетках крови.
У пациента не было инфекционных осложнений, признаков РТПХ не было. Полный химеризм несколько раз подтверждался анализом группы крови. До настоящего времени (через 19 месяцев после ТКМ) у пациента не было инфекции, и не было осложнений, связанных с отсроченной трансплантацией.
Иммунологические исследования
Иммунологические результаты до и после BMT суммированы в таблице 2.
Таблица 2b Иммунологические исследования: in vitro Функция Т-лимфоцитов (индекс стимуляции)Как характерно для дефицита PNP, наблюдались серьезные нарушения Т- и В-клетки функционируют до ТКМ с низким количеством как Т-, так и В-клеток. Однако абсолютный уровень иммуноглобулинов был нормальным. На +95 день после BMT было обнаружено, что Т-клетки были нормальными по количеству и распределению субпопуляций и показали регулярную пролиферацию после стимуляции in vitro .